Сборка генома de novo
Задание 1
Дано:
8254632 коротких чтений бактерии Buchnera aphidicola, полученных по технологии Illumina (SRR4240360)
Buchnera aphidicola - симбиотическая бактерия тлей. Так как тли питаются растительным соком, в их рационе не хватает некоторых аминокислот. Некоторые из них (незаменимые аминокислоты) тли не могут синтезировать сами, поэтому это осуществляет симбиотическая бактерия Buchnera aphidicola. Тля, в свою очередь, обеспечивает бактерию энергией, углеродом и азотом. Размер генома Buchnera aphidicola достаточно мал - состаляет 628164 bp (CP009253.1). Это связано с тем, что из-за симбиотического образа жизни бактерия утратила многие гены, однако в ней обнаружены гены синтеза тех аминокислот, которые не способна синтезировать тля. [1]
Рис. 1. Тля и симбиотическая бактерия Buchnera aphidicola
Подготовка чтений
С помощью программы trimmomatic были удалены остатки адаптеров:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 SRR4240360.fastq noadapt.fastq ILLUMINACLIP:adapters.fasta:2:7:7
Далее были удалены нуклеотиды с низким качеством с концов чтений, причем были оставлены чтения длиной не менее 30:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 noadapt.fastq outfile.fastq TRAILING:20 MINLEN:30
В процессе очистки было удалено 269878 (3,27%) чтений. Таким образом, после очистки из 8254632 осталось 7984754 чтений (96,73%).
Сборка генома (Velvet)
После этого была использована программа Velvet. Она осуществляет сборку генома из коротких последовательностей с помощью графов де Брайна. Этот алгоритм позволяет избавиться от проблемы повторов, возникающей при использовании альтернативного алгоритма сборки геномов overlap-layout-consensus (OLC).
Алгоритм де Брайна можно разделить на два этапа:
1. Cоставление слов определенной длины (k-меров) из коротких ридов с последующим созданием из них графа. Важно, что число k должно быть нечетным, чтобы избежать палиндромов, а также меньше чем длина ридов. В нашем случае риды были разбиты на k-меры длины k=29 и k=25 (чтения короткие и не парные - short, assem - директория для выходных файлов):velveth assem 29 -short -fastq outfile.fastq velveth assem2 25 -short -fastq outfile.fastq
2. Сборка контигов из составленного графа. Cборка на основе k-меров была проведена с помощью программы velvetg (assem, assem2 - директории с файлами, полученными на предыдущей стадии):
velvetg assem velvetg assem2
В результете было получено несколько файлов с информацией о сборках:
Замечение: Во всех этих файлов длины указаны для k-меров. чтобы определить длину контигов в bp нужно прибавить значение (k-1). Далее результаты представлены с длинами, указанными в файлах ((k-1) не прибавлялось!).
Анализ сборок c k=25 и k=29
Для анализа сборок был взят файл contigs.fa, т.к. он не содержит ненужной информации о контигах с длинами 1, как файл stats.txt. Так как файл contigs.fa содержит последовательности в fasta-формате, то сначала для удобства дальнейшей работы с помощью скрипта из него был получен файл с длинами контигов и их покрытиями.
Параметры сборки для k=25 и k=29 получились отличными друг от друга(табл. 1, 2). Во-первых, у сборки с k=25 число контигов почти в 2 раза больше, чем для сборки с k=29. Во вторых, общая длина контигов у сборки с k=25 больше на 144087. Однако показатель N50 (наибольшее число такое, что контигами длины > N50 покрыто 50% генома) у этой сборки гораздо ниже, то что говорит о том, что качество сборки лучше при k=29.
Что касается длин контигов и их покрытий, то у сборки с большим k-мером длины контигов тоже больше, однако их покрытие ниже. У сборки с более низким значением k длина самого большого контига почти в 4 раза меньше, чем у сборки с большим значением k. Однако покрытие больше в 1.75 раза.
| Значение k | N50 | Общая длина контигов | Количество контигов |
| k=29 | 67095 | 707483 | 1131 |
| k=25 | 5627 | 851570 | 2276 |
| k=29 | k=25 | ||||
| № | Длина | Покрытие | № | Длина | Покрытие |
| 1 | 94956 | 43.695322 | 1 | 26018 | 74.528980 |
| 2 | 70305 | 49.381367 | 2 | 23552 | 70.151664 |
| 3 | 70300 | 42.029132 | 3 | 21040 | 62.380038 |
На рис. 2 представлены графики, отражающее количество контигов(длина которых больше 25 или 29) в зависимости от покрытия для k=25 и k=29. Анлизируя полученные результаты можно сделать вывод о том, что более длинные k-меры обеспечивают специфичность (меньше вероятность того, что наложатся неправильные последовательности), но меньшее покрытие.
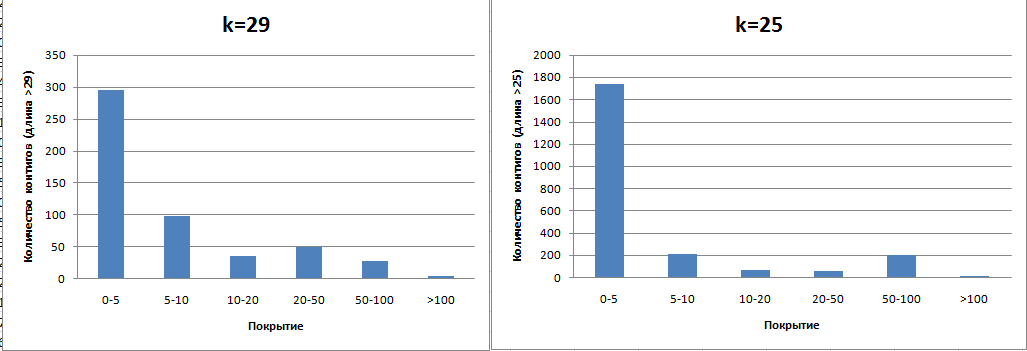
Были также обнаружены аномальные покрытия (для сборки с k=29):
Выравнивание полученных контигов с хромосомой Buchnera aphidicola
Характеристики выравнивания megablast самых длинных контигов (сборка с k=29) с хромосомой Buchnera aphidicola (GenBank/EMBL AC — CP009253) представлены в табл.3.
| Параметр | Контиг №1 (94956 bp) | Контиг №2 (70305 bp) | Контиг №3 (70300 bp) |
| Координаты участка хромосомы, соответствующего контигу | 467412 - 474667 | 5124 - 44693 | 5124 - 44693 |
| Число однонуклеотидных различий | 7181 | 9503 | 10494 |
| Число гэпов | 208 | 130 | 390 |
То же самое было проделано с контигом №946 (его покрытие 116,891891), однако megablast не нашел соответствий между последоваетльностью этого контига и хромосомой Buchnera aphidicola .
Сборка с помощью SPAdes
SPAdes (St. Petersburg genome assembler) - это программа, разработанная для сборки небольших геномов, например, бактерий. Сборка также основана на построении графа де Брайна, однако значение k подбирается программой автоматически в зависимости от длины ридов и типа базы данных. SPAdes был запущен со следующими параметрами:
spades.py --only-assembler --careful -s outfile.fastq -o spades
В результате было получено несколько файлов: contigs.fasta, contigs.fastg, scaffolds.fasta и scaffolds.fastg. Далее из файла contigs.fasta был получен файл out с основной информацией о контигах с помощью небольшого скрипта на Python.
| Значение k | N50 | Общая длина контигов | Количество контигов |
| 33 | 78297 | 802000 | 404 |
| № | Длина | Покрытие |
| 1 | 174223 | 17,2028 |
| 2 | 104293 | 16,111 |
| 3 | 91646 | 15,6451 |
Сравнивая результаты сборки SPAdes с результатами сборки Velvet(k=29) можно обнаружить следующие различия:
Для самого большого контига было запущено выравнивание megablast с хромосомой Buchnera aphidicola . Результаты приведены в табл. 6.
| Параметр | Контиг №1 (174223 bp) |
| Координаты участка хромосомы, соответствующего контигу | 532851 - 550219 |
| Число однонуклеотидных различий | 3235 |
| Число гэпов | 443 |
Из всего вышесказанного можно сдедлать вывод о том, что сборка с помощью программы Spadex более качественнее, чем с помощью Velvet с параметрами k=29 и k=25. К тому же, Spadex осуществляет сборку скаффлодов из полученных контигов. Однако время работы Spadex значительно больше времени работы Velvet.
Сборка с использованием половины исходных ридов
Из файла, полученного после очистки чтений (outfile.fastq) была удалена половина последовательностей - в итоге в файле осталось 3992378 чтений. На основе полученного файла (outfile_half.fastq) проводилась сборка с помощью программы Velvet.
velveth half 29 -short -fastq outfile_half.fastq velvetg half
| N50 | Общая длина контигов | Количество контигов |
| 16037 | 672510 | 266 |
Оказалось, что у данной сборки N50 значительно ниже, чем у сборки со всеми ридами (16037 против 67095).Количество контигов(длина которых больше 29) также значительно ниже (266 против 1131). Однако общая длина контигов примерно одинаковая.
Что касается покрытия и длин самых больших контигов (табл.8), то они тоже меньше, чем у сборки со всеми ридами. В частности, покрытие оказалось меньше почти в 2 раза.
| № | Длина | Покрытие |
| 1 | 90828 | 22,815157 |
| 2 | 32734 | 24,76025 |
| 3 | 29248 | 23,504854 |
Таким образом, чем больше ридов дано изначально, тем лучше качество сборки: больше значение N50, покрытие и длины самих ридов.