|
||||||||||||||||||||||||||||||||
|
|
Структурные гомологи Cu,Zn-супероксиддисмутазы 1PU0С помощью сервиса PDBeFold было найдено четыре гомолога белка Cu,Zn-супероксиддисмутазы человека. Поиск осуществлялся по цепи A из PDB-структуры 1PU0. В таблице 1 представлена краткая характеристика найденных гомологов. На рисунке 1 изображено наложение структур гомологов на структуру 1PU0 (методами отображения ribbon или cartoon).
* - в скобках указана доля, которую составляет длина выравнивания от длины исходного белка 1PU0 (153 а.о.) 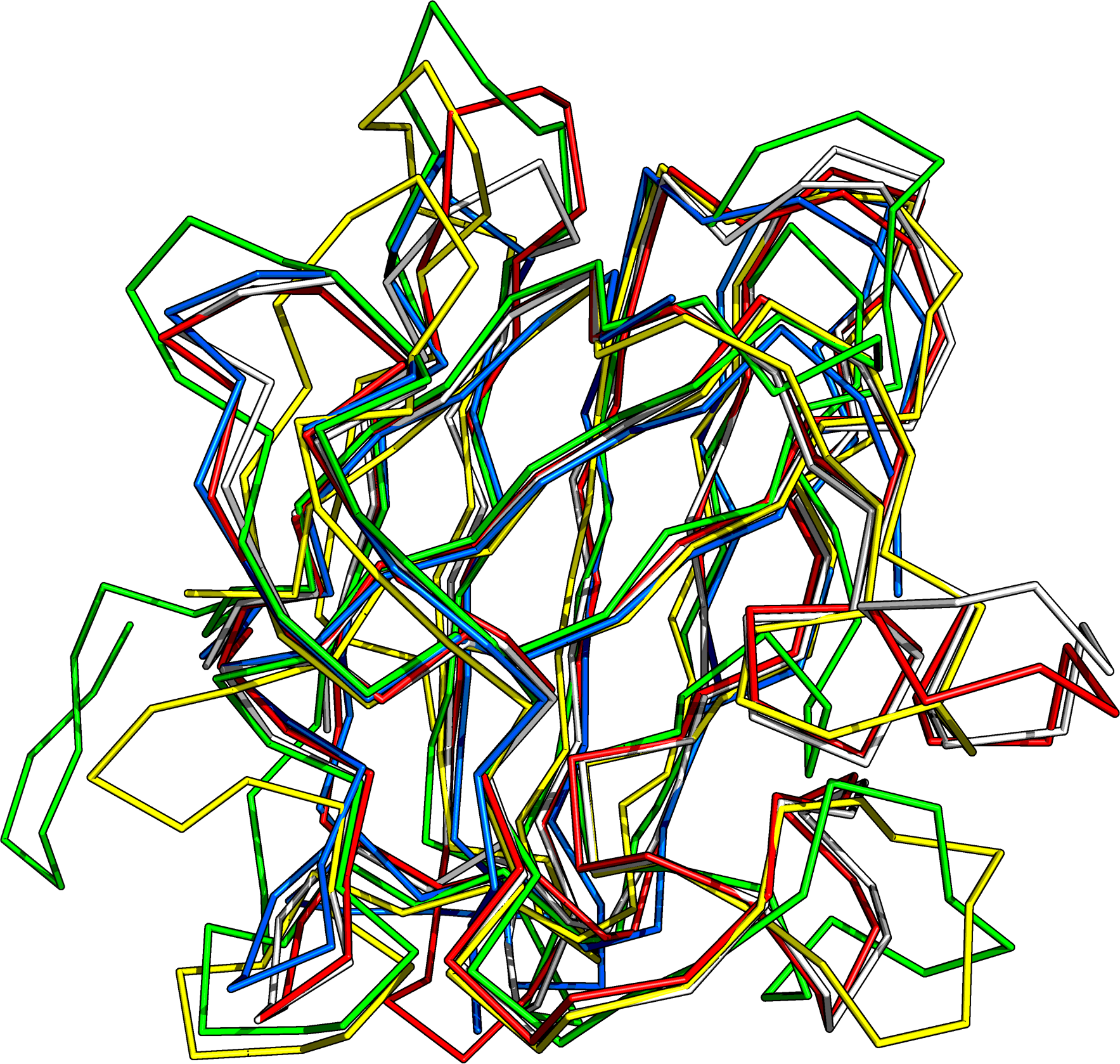
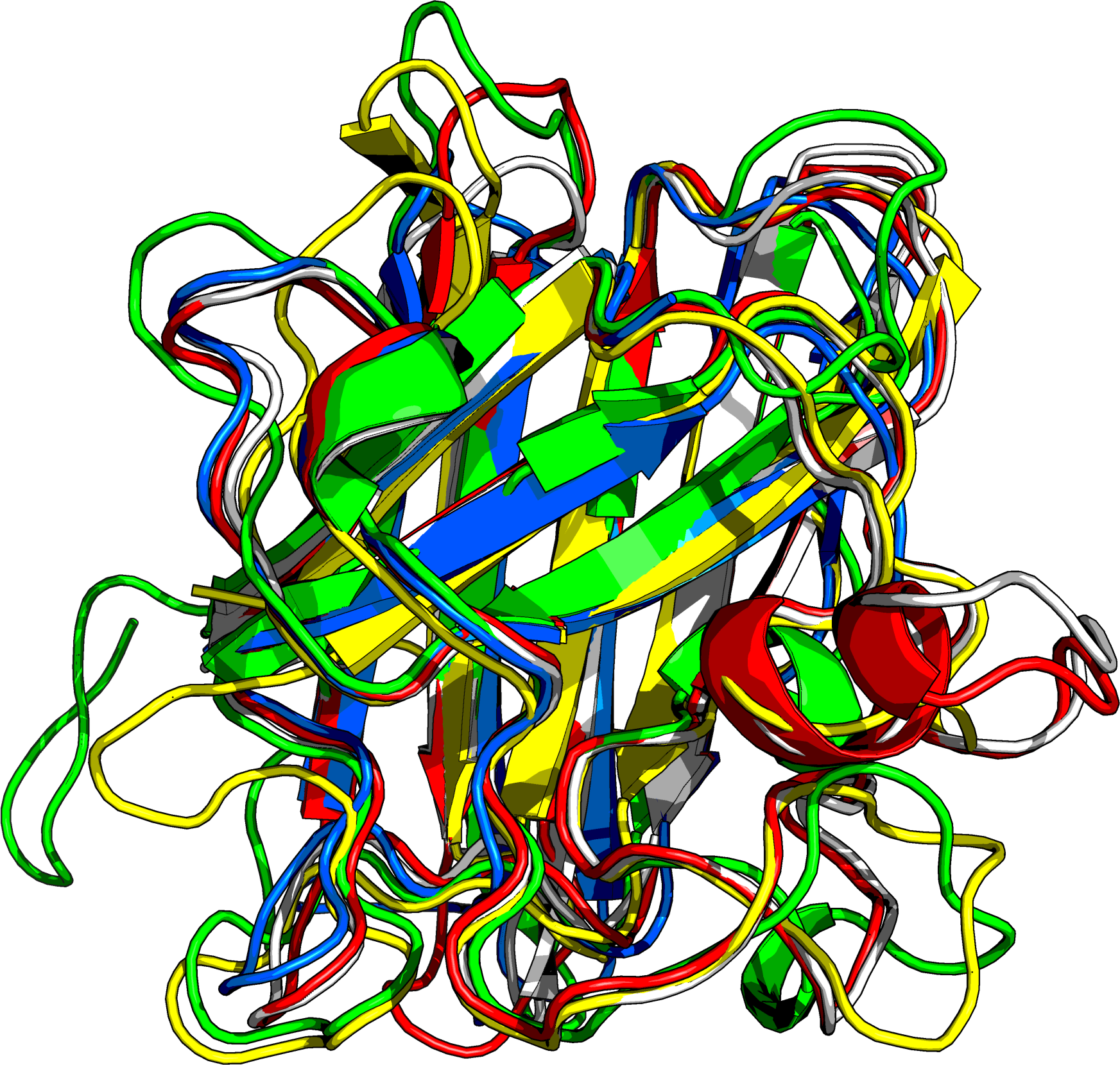
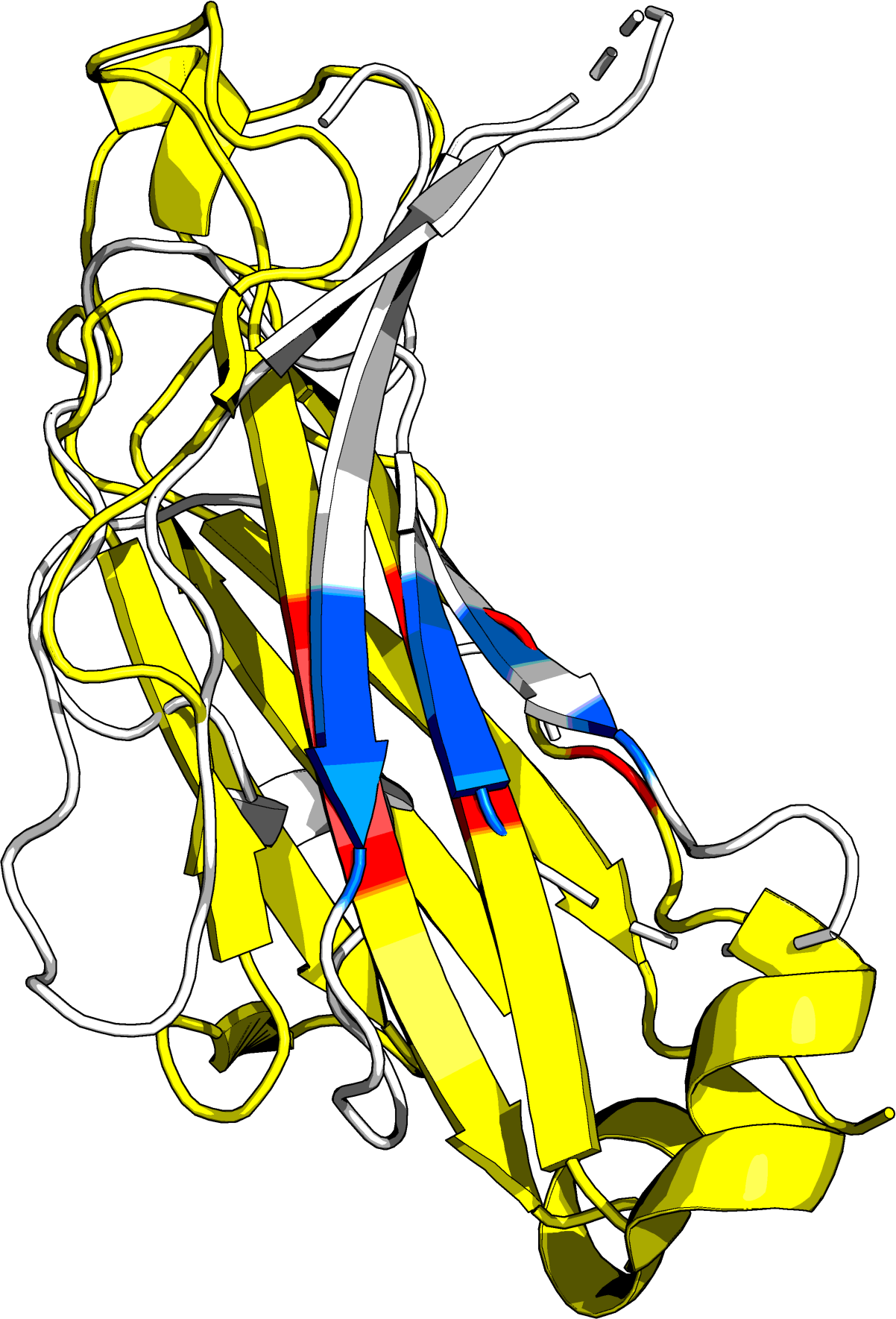
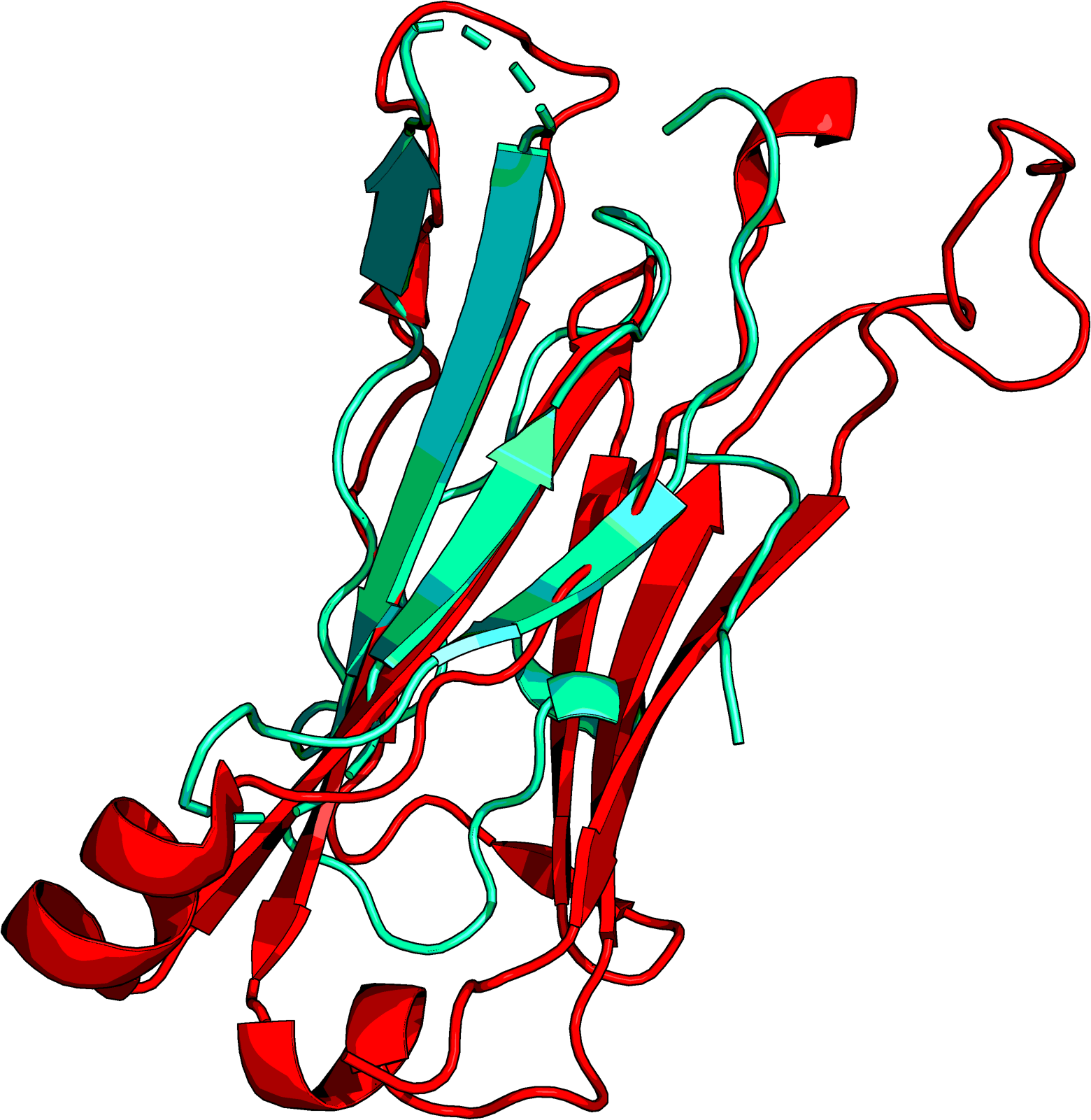
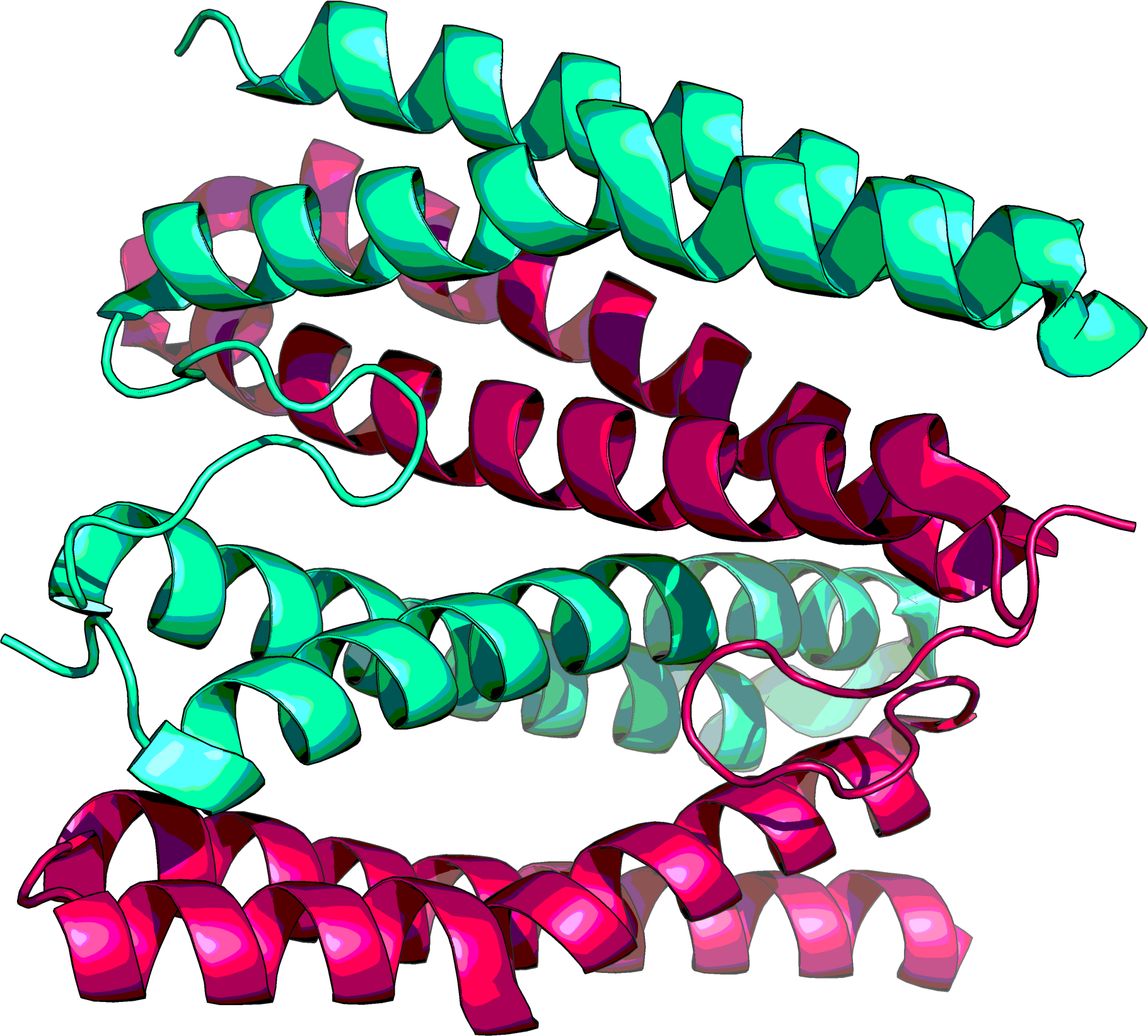
Рисунок 1. Совмещение структур Cu,Zn-СОД (1PU0, красная цепь) со структурами гомологов из разных организмов: Hydra vulgaris (4OJA, белая цепь), Megavirus chilensis (4U4I, синяя цепь), Bacillus subtilis (1S4I, желтая цепь) и Candida albicans (4N3U, зеленая цепь). Представлены изображения по ribbon (слева) и cartoon (справа).
Как видно из рисунка 1, структуры белков достаточно хорошо согласуются и накладываются друг на друга. Основные
отличия наблюдаются в участках петель.

Рисунок 2. Выравнивание по структуре. Аминокислоты покрашены по стандартной схема AliView. 
Рисунок 3. Выравнивание программой Muscle с настройками по умолчанию.
В полученных выравниваниях есть некоторые отличия. На рисунке 4a представлен один из таких участков выравнивания,
а на рисунке 4b - соответствующие аминокислотные остатки в наложении структур.


Рисунок 4a. Участок с несоответствием выравнивания по структурам (слева, позиция 70) выравниванию по последовательности (справа, позиция 61). 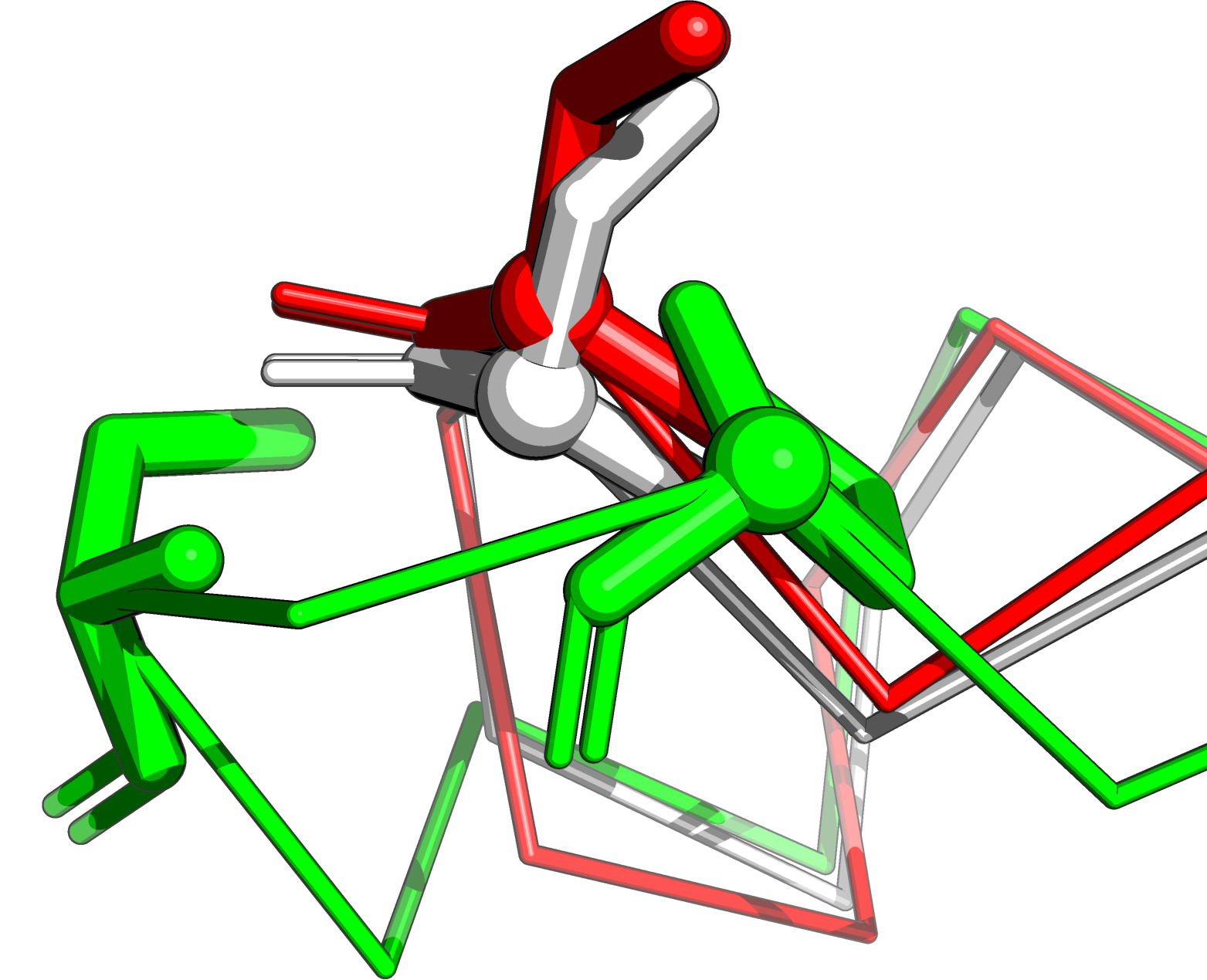
Рисунок 4b. Положение аминокислот из 70 колонки структурного выравнивания в наложении структур. Показаны структуры 1PU0 (красная цепь), 4OJA (белая цепь) и 4N3U (зеленая цепь). Сферами отмечены Cα-атомы аминокислот из одной колонки выравнивания. Отдельно отмечен остаток Ser84, помещенный в одну колонку с Ser53 и Thr54 программой Muscle. Еще один пример - позиция 136 в структурном выравнивании и соответствующая ей позиция 118 выравнивания по последовательностям. В этой позиции для последовательности 1S4I стоит Leu или Ser (рисунок 5a). Однако в этом случае не так просто определить, какое из выравниваний более правильное. На рисунке 5b представлено положение описываемых остатков в структуре (желтая структура) по сравнению с 1PU0 (красная структура). Видно, что расстояния между Cα-атомами остатков His из 1PU0 и Leu или Ser из 1S4I составляет 2.5 и 4.5 Å, соответственно. Следовательно, Leu более близок по структуре к другим остаткам из 136 колонки выравнивания. Однако по свойствам аминокислот функционально в этой позиции более подходящим будет Ser, а Leu вполне может располагаться в выпетливании, возникшем из-за вставки в последовательность 1S4I. 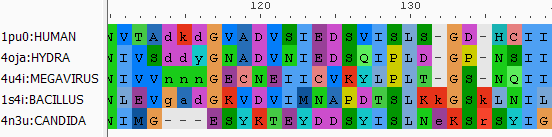
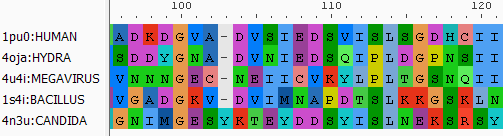
Рисунок 5a. Участок с несоответствием выравнивания по структурам (сверху, позиция 136) выравниванию по последовательности (снизу, позиция 118). 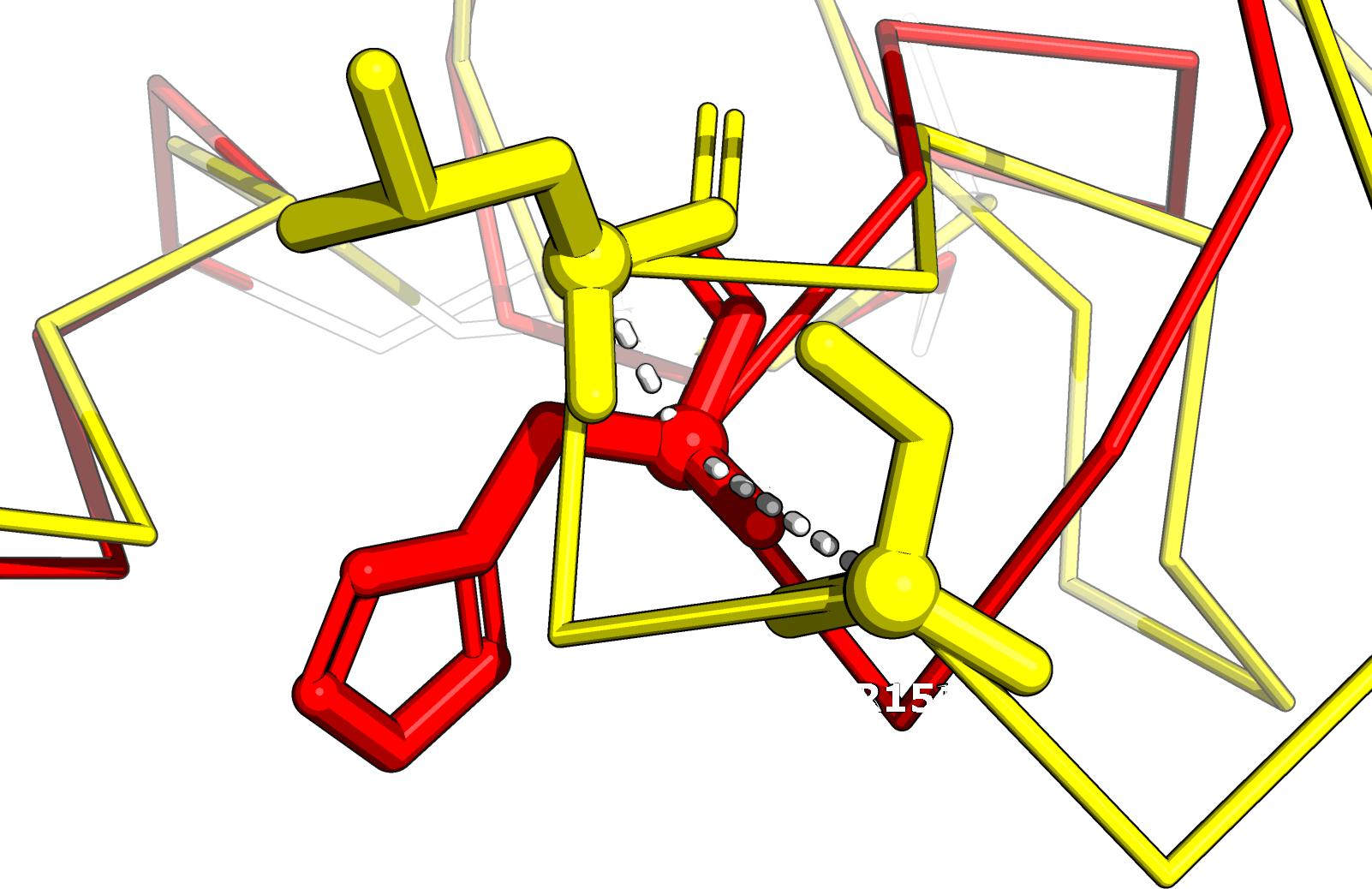
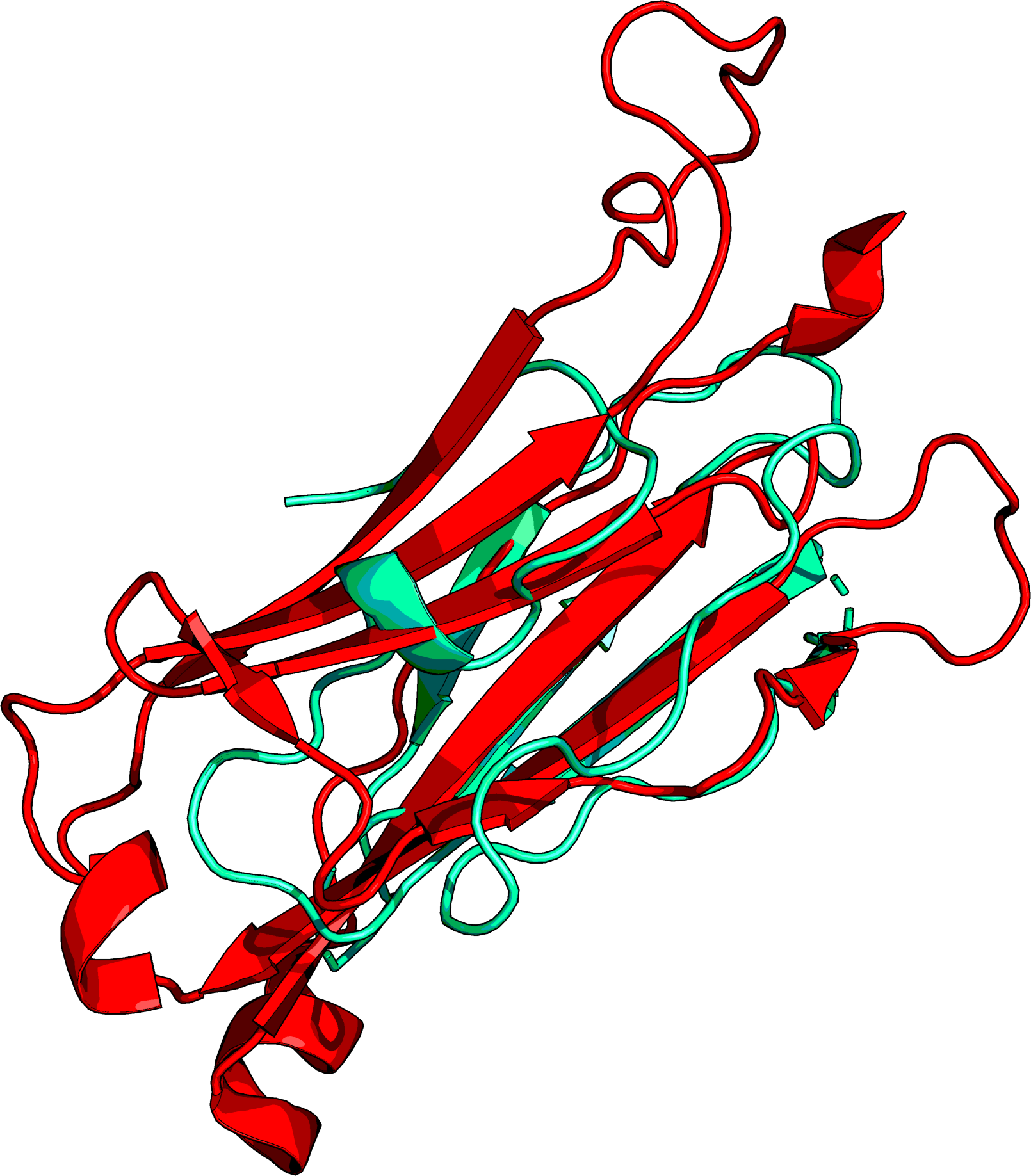
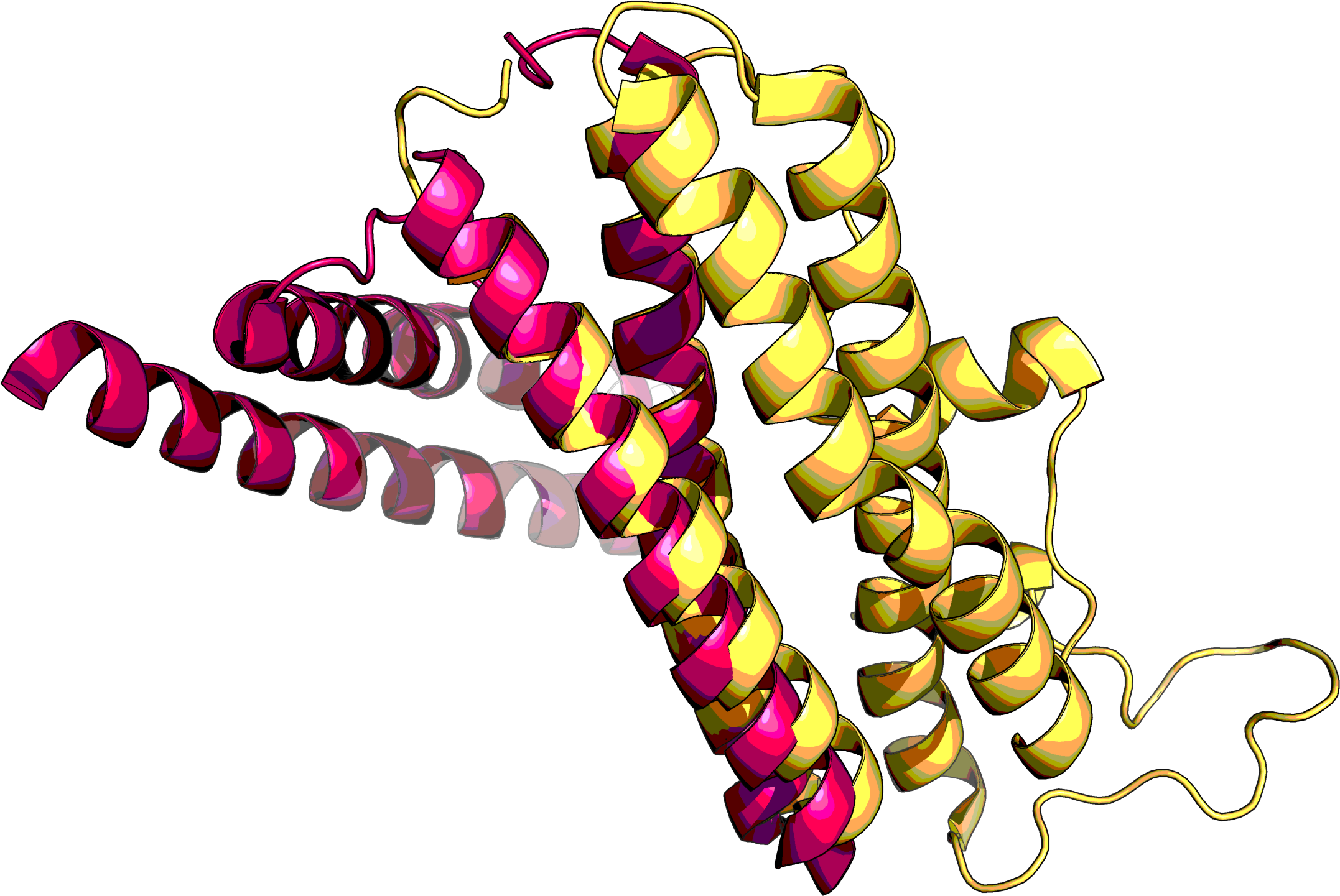
Рисунок 5b. Положение аминокислот из 136 колонки структурного выравнивания в наложении структур. Показаны структуры 1PU0 (красная цепь) и 1S4I (желтая цепь). Сферами отмечены Cα-атомы аминокислот. Также отмечены расстояния между Cα-атомами остатков His из 1PU0 и Leu или Ser из 1S4I. Совмещение доменов Т-клеточного рецептора по заданному выравниванию. SheePДля выполнения этого задания было выбрано два домена: участок структуры D:118-203 из 1BD2 (цепь α) и E:119-246 из 1QSE (цепь β). Выбранные структуры и домены изображены на рисунках 6a и 6b. На рисунке 7 показано совмещение доменов в PyMOL (командой align). 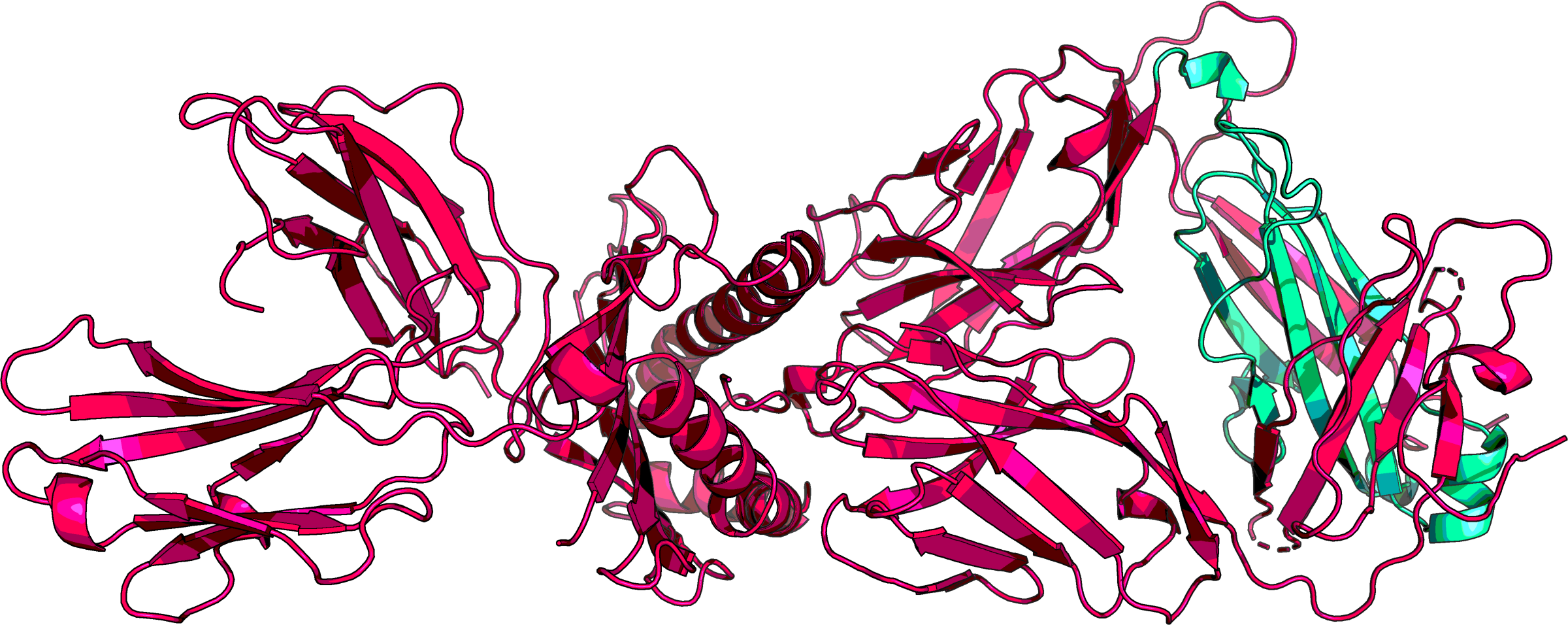
Рисунок 6a. Структура 1BD2. Цветом выделен участок D:118-203 - домен α-цепи Т-клеточного рецептора. 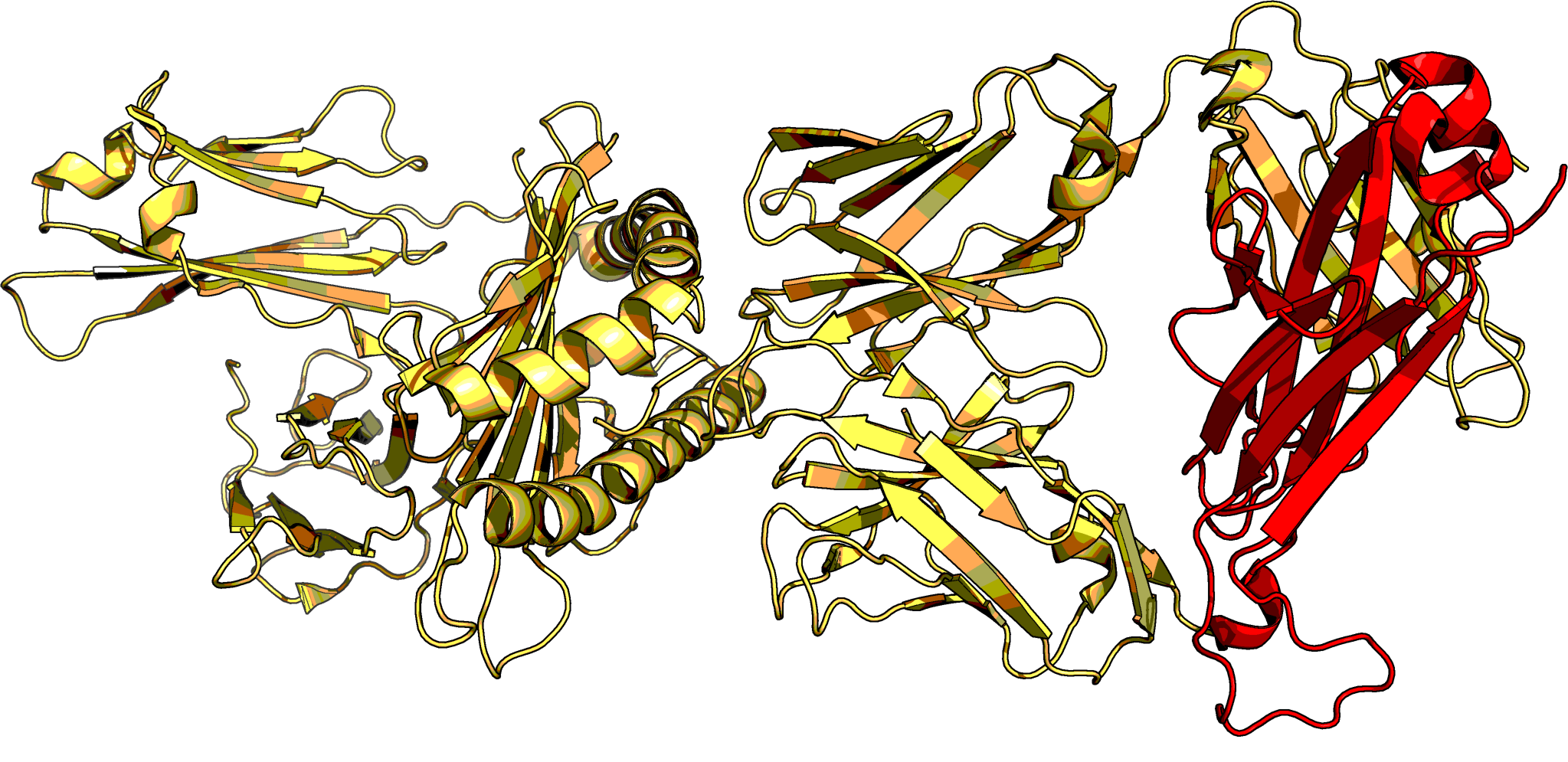
Рисунок 6b. Структура 1QSE. Цветом выделен участок E:119-246 - домен β-цепи Т-клеточного рецептора. 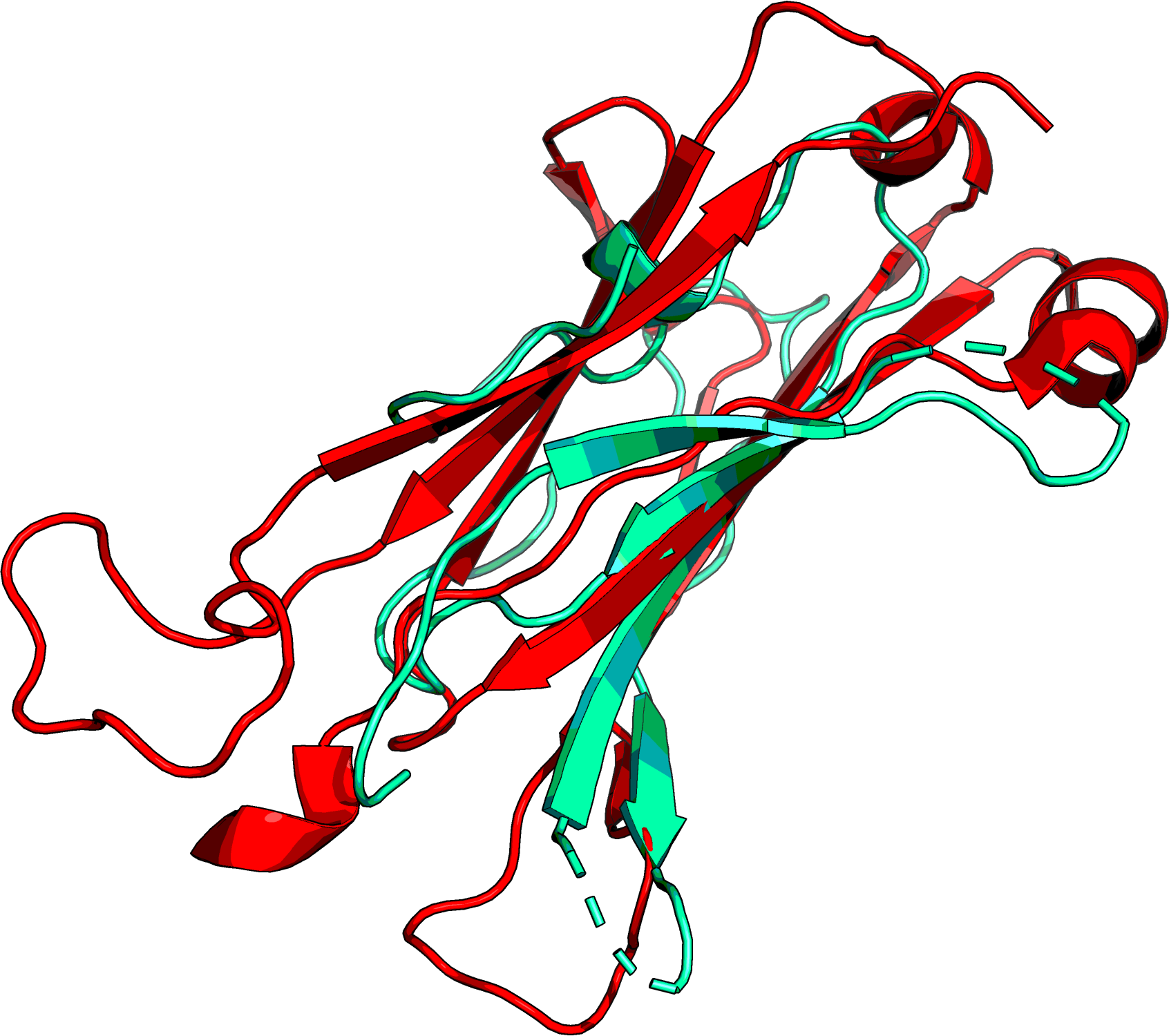
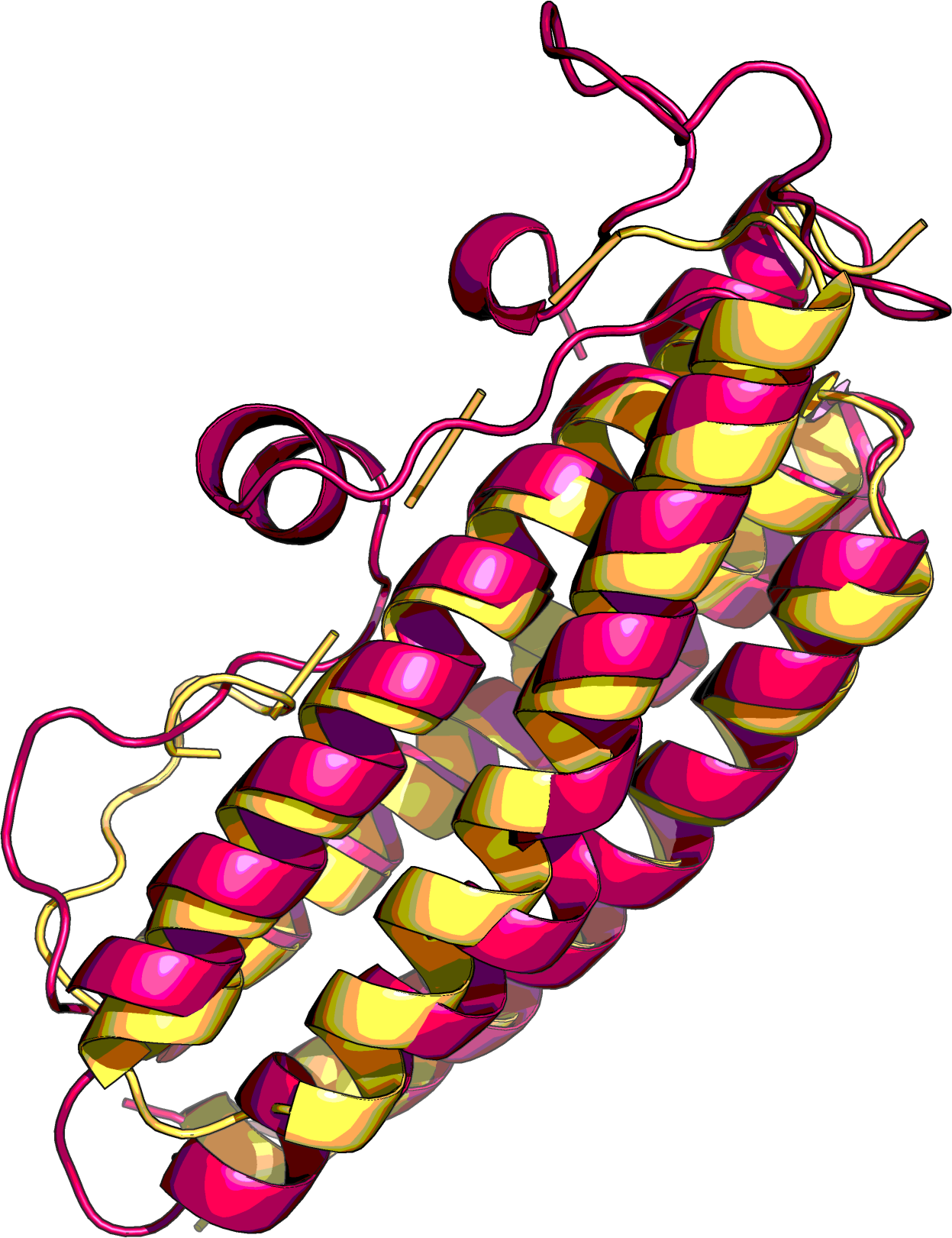
Рисунок 7. Совмещение α-цепи (сине-зеленая) и β-цепи (красная) командой align в PyMOL. Затем с помощью программы SheeP были получены карты β-листов для выбранных доменов. Карты представлены на рисунках 8a и 8b (для α-цепи и β-цепи, соответственно). В каждой карте был найден консервативный остаток цистеина: Cys139 в цепи α и Cys147 в цепи β. Эти остатки были использованы для построения выравнивания доменов. 
Рисунок 8a. Карта β-листов для α-цепи Т-клеточного рецептора. 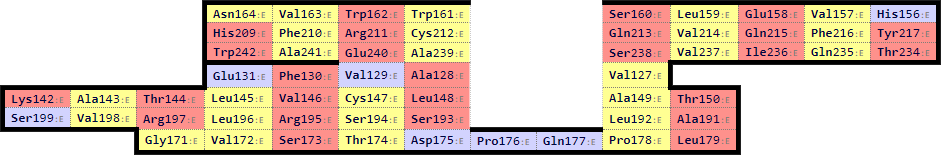
Рисунок 8b. Карта β-листов для β-цепи Т-клеточного рецептора. Совмещение структур производилось по β-листу, содержащему консервативный цистеин. Использованные команды:
|
select alpha, 1bd2 and chain D and resi 126+128+138-140+179-181 and name CA
select beta, 1qse and chain E and resi 128+130+146-148+193-195 and name CA pair_fit alpha, beta | ||||||||||||||||||||||||||||||