Построение деревьев по нуклеотидным последовательностям. Паралоги
Построение филогенетического дерева на основании 16S рибосомальной РНК
Для получения последовательностей 16S рибосомальной РНК выбранных мною бактерий был написан скрипт на bash. Для замены имен бактерий на мнемоники им используется скрипт на Python и таблица соответствий имен и мнемоник. Параметры были установлены так, чтобы для каждой бактерии скачивать только одну ($THRESHOLD_to_FILE=1) последовательность 16S РНК из одного ($THRESHOLD=1) штамма.
Далее полученный файл с последовательностями mnem_total.16srna был выравнен сервисом MUSCLE. Файл с выравниванием. Далее с помощью программы MEGA с использованием метода Maximum Likelihood. Полученный файл в формате .nwk. Полученное дерево изображено на рис. 1.
Полученное дерево и "настоящее дерево" (рис. 2) сильно отличаются. К примеру, в дереве, построенном по 16S РНК есть ветвь {GEOKA, CLOB1} vs {LISMO, STAA1, ENTFA, LACLM, STRP1, STRPN}, в то время как на настоящем дереве такая ветвь отсутствует. Получаем, что использование 16S РНК в данном случае не дает достоверного филогенетического дерева. Сравнение деревьев было проведено при помощи скрипта на python. Результаты представлены в таблице 1.
| Имя первого дерева | Имя второго дерева | Расстояние|Максимальное расстояние | Разбиения, которые есть в первом дереве, но нет во втором | Разбиения, которые есть во втором дереве, но нет в первом |
| 16s_rib_max_lik.nwk | natural_tree.nwk | 4|16 | {'CLOB1', 'GEOKA'} vs {'ENTFA', 'LACLM', 'LISMO', 'STAA1', 'STRP1', 'STRPN'} ||| {'CLOB1', 'ENTFA', 'GEOKA', 'LACLM', 'STRP1', 'STRPN'} vs {'LISMO', 'STAA1'} | {'CLOB1', 'GEOKA'} vs {'ENTFA', 'LACLM', 'LISMO', 'STAA1', 'STRP1', 'STRPN'} ||| {'CLOB1', 'ENTFA', 'GEOKA', 'LACLM', 'STRP1', 'STRPN'} vs {'LISMO', 'STAA1'} |
| natural_tree.nwk | 16s_rib_max_lik.nwk | 4|16 | {'CLOB1', 'GEOKA'} vs {'ENTFA', 'LACLM', 'LISMO', 'STAA1', 'STRP1', 'STRPN'} ||| {'CLOB1', 'ENTFA', 'GEOKA', 'LACLM', 'STRP1', 'STRPN'} vs {'LISMO', 'STAA1'} | {'CLOB1', 'GEOKA'} vs {'ENTFA', 'LACLM', 'LISMO', 'STAA1', 'STRP1', 'STRPN'} ||| {'CLOB1', 'ENTFA', 'GEOKA', 'LACLM', 'STRP1', 'STRPN'} vs {'LISMO', 'STAA1'} |
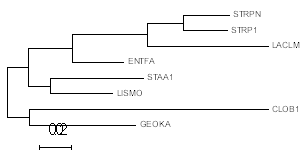
Рис. 1. Дерево 16S-рибосомных РНК построенное с помощью программы MEGA.
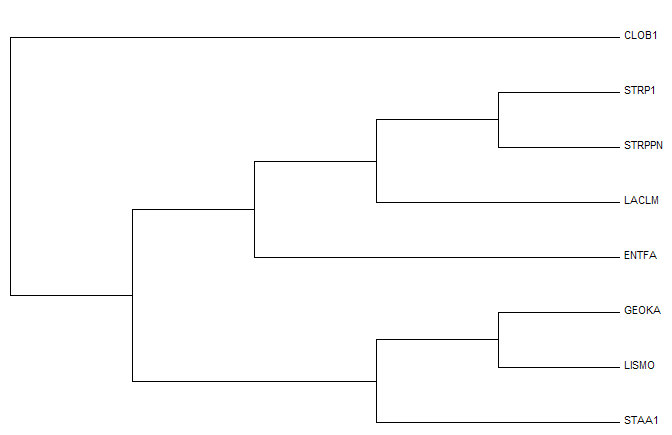
Рис. 2. Филогенетическое дерево бактерий с использованием метода Maximum Likelihood.
Построение и анализ дерева, содержащего паралоги
Протеомы отобранных мною бактерий были слиты в один файл (на сайте файл представлен в архиве) total.fasta.zip. Затем была из него была создана база белков при помощи команды:
makeblastdb -in total.fasta -dbtype prot
Затем последовательность белка CLPX_BACSU P50866.fasta была отбластована на эту базу при помощи команды:
blastp -query P50866.fasta -db total.fasta -evalue 0.001 -outfmt 7 -word_size 2 -out homologs_exp.txt
В результате был получен файл с хитами с порогом e-value=0.001 - homologs_exp.txt. Далее был получен список USA my_id.bs найденных гомологов при помощи команды:
cat homologs.txt | egrep -v '#' | awk ' {print $2}' | awk ' BEGIN {FS = "|"} {print "fasta::total.fasta:" $3}' | sort -u > my_id.bs
Затем был получен итоговый файл с последовательностями всех гомологов homologs.fasta был получен при помощи команды:
seqret @my_id.bs homologs.fasta
С помощью скрипта на Python был получен файл последовательностей, с полями '>...', содержащими только названия белков - homologs_mnem.fasta
Последовательности был выравнены при помощи сервиса MUSCLE (homologs_aln.fasta - файл выравнивания) и на основании полученного выравнивания с использованием метода Maximum Likelihood было построено дерево, изображение которого приведено на рис.3 (файл дерева - homologs_tree.nwk).
Образованию новых белков в результате видообразования соответствует множество {CLPX_GEOKA, CLPX_LISMO} (выделено красным цветом). Паралогами с большой долей верочтности являются белки A7FYT8_CLOB1 и A7FZB1_CLOB1 (выделено синим цветом).
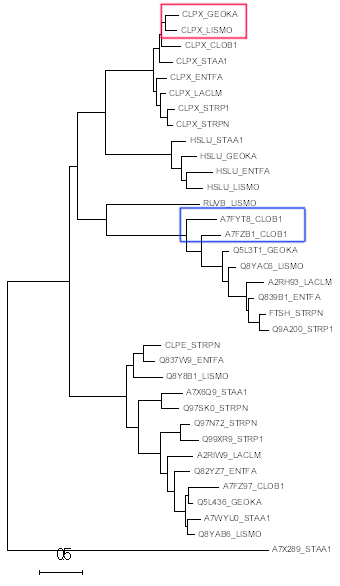
Рис. 3. Филогенетическое дерево бактерий с использованием метода Maximum Likelihood.
