Задание 1. Введение
В этом практикуме я работал со структурами 6TJ3 и 6TJ4. Это структуры N-терминального домена основной легкой цепи Plasmodium falciparum (P. falciparum essential light chain, PfELC), полученные методами ЯМР и РСА, соответственно1.
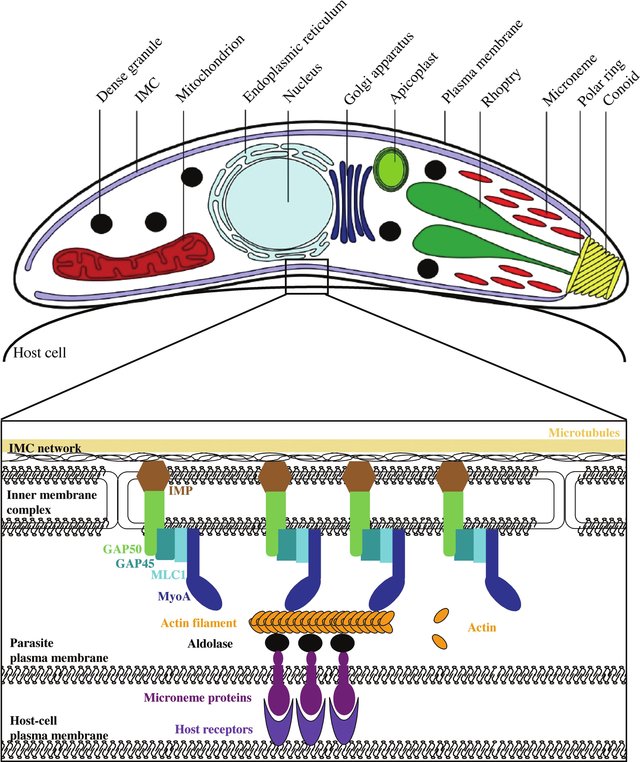
PfELC - это компонент глайдосомы данного вида простейших. Дело в том, что представители типа Apicomplexa обладают уникальным типом движения - скольжением (gliding), которое позволяет им без труда перемещаться в тканях хозяина. На рисунке 1 изображена модель глайдосомы Toxoplasma gondii - органеллы, позволяющей перемещаться этому паразиту семейства кошачьих.
PfELC в данном комплексе связывается с миозином A, предположительно приводя к изменению вторичной структуры последнего, однако точная роль данного белка пока неизвестна.
Поскольку исследователи получили структуры данного белка как с помощью ядерно-магнитного резонанса (ЯМР), так и с помощью РСА (ретнгенструктурного анализа), у нас есть возможность сравнить их между собой.
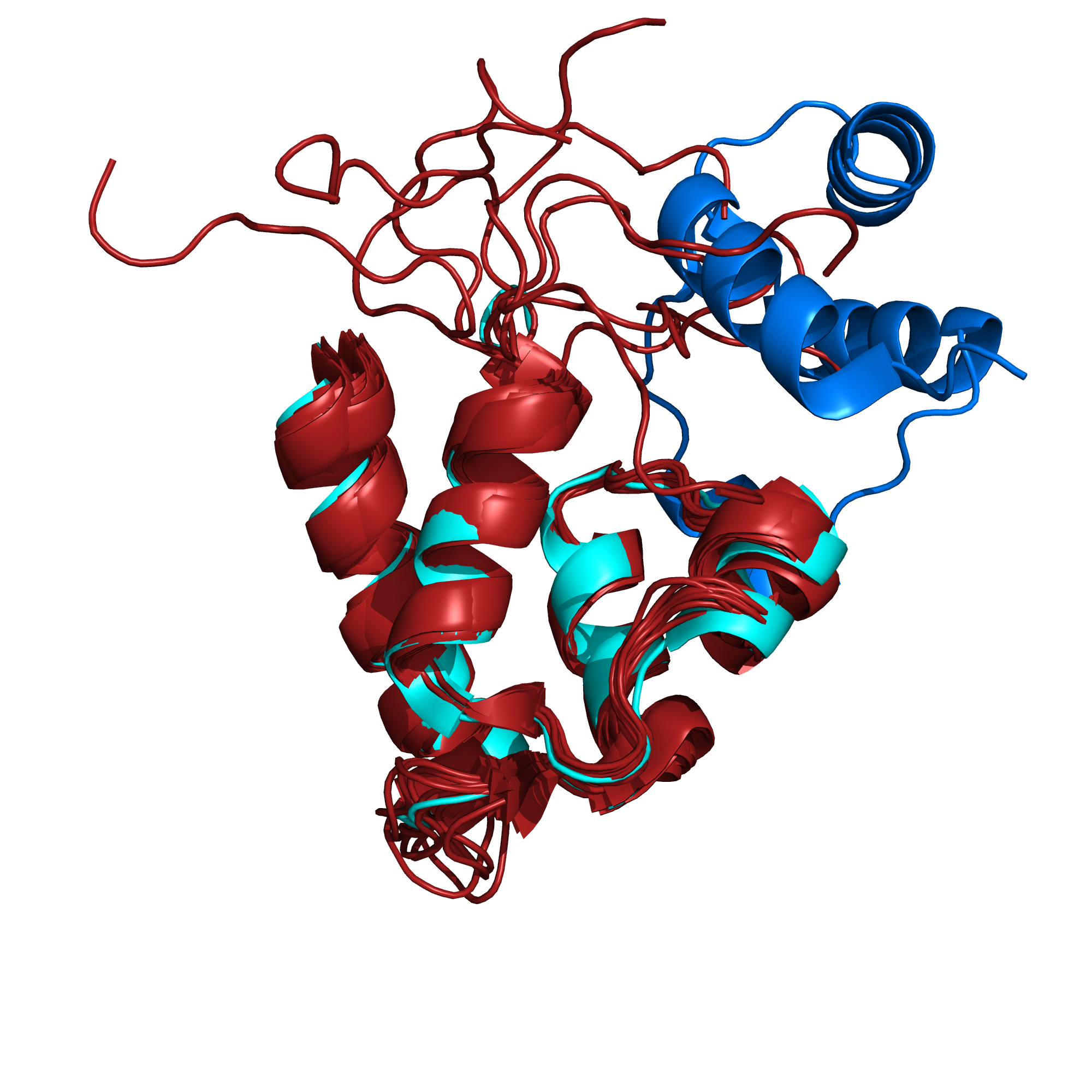
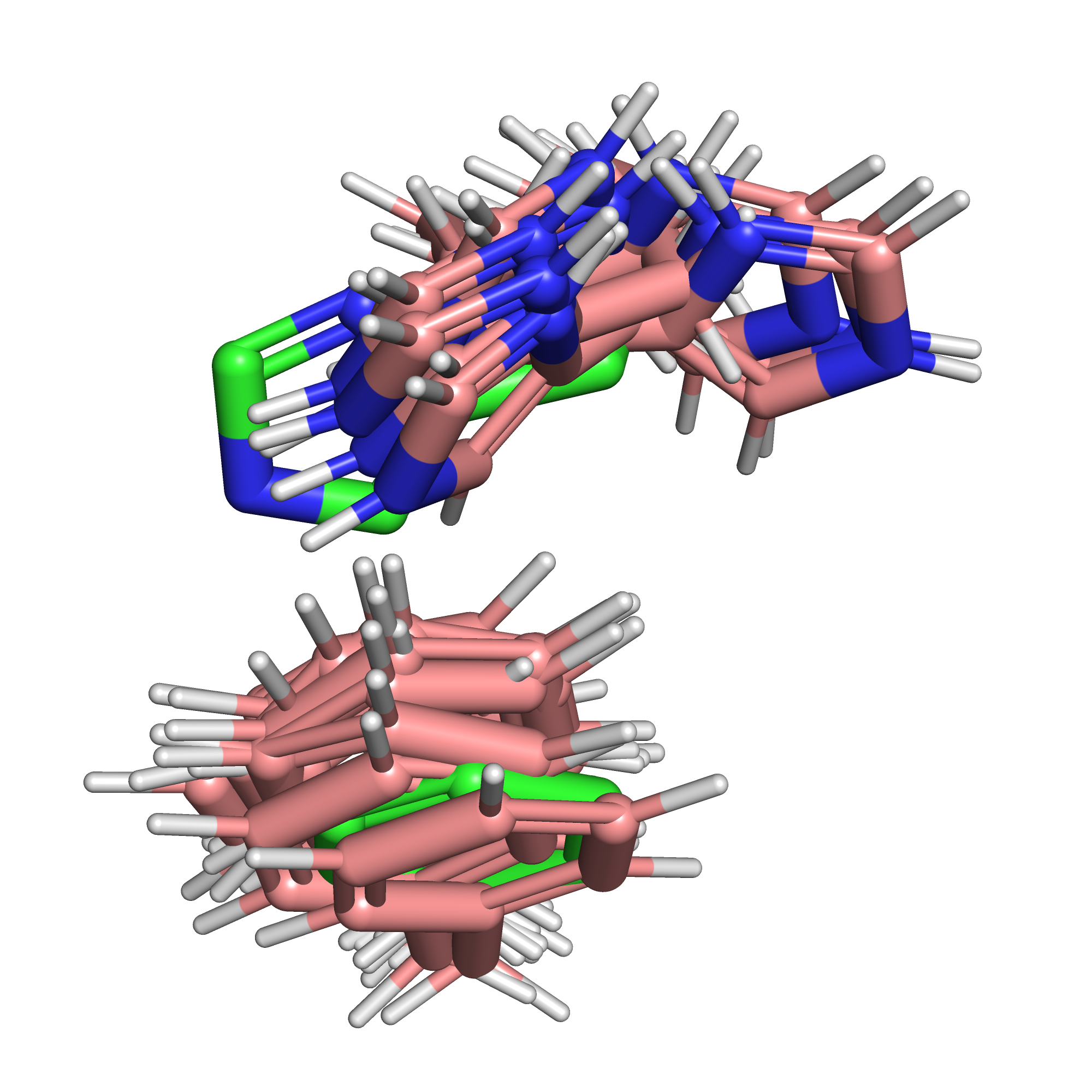
На рисунке 2 изображены две структуры в целом. Сразу заметно, что с помощью ЯМР получают не один единственный вариант структуры, а несколько разных, поскольку по набору ограничений на расстояния структуру можно восстановить по-разному. В данном случае авторы получили ансамбль из 100 конформеров, из которых выложили 10. Для структуры, полученной РСА, важным параметром является разрешение: в данном случае оно равно 1.5Å (полнота данных - 99%). Кроме того, РСА-структура содержит две цепи, а ЯМР - только одну, что также отражено на рис. 2.
В целом, основная часть данного домена очень похожим образом определена как ЯМР, так и РСА. Главное, чем серьезно различаются конформеры ЯМР - это C-концевой фрагмент, который в РСА-структуре имеет самые высокие значения B-фактора, что говорит о его подвижности (рис. 3.)
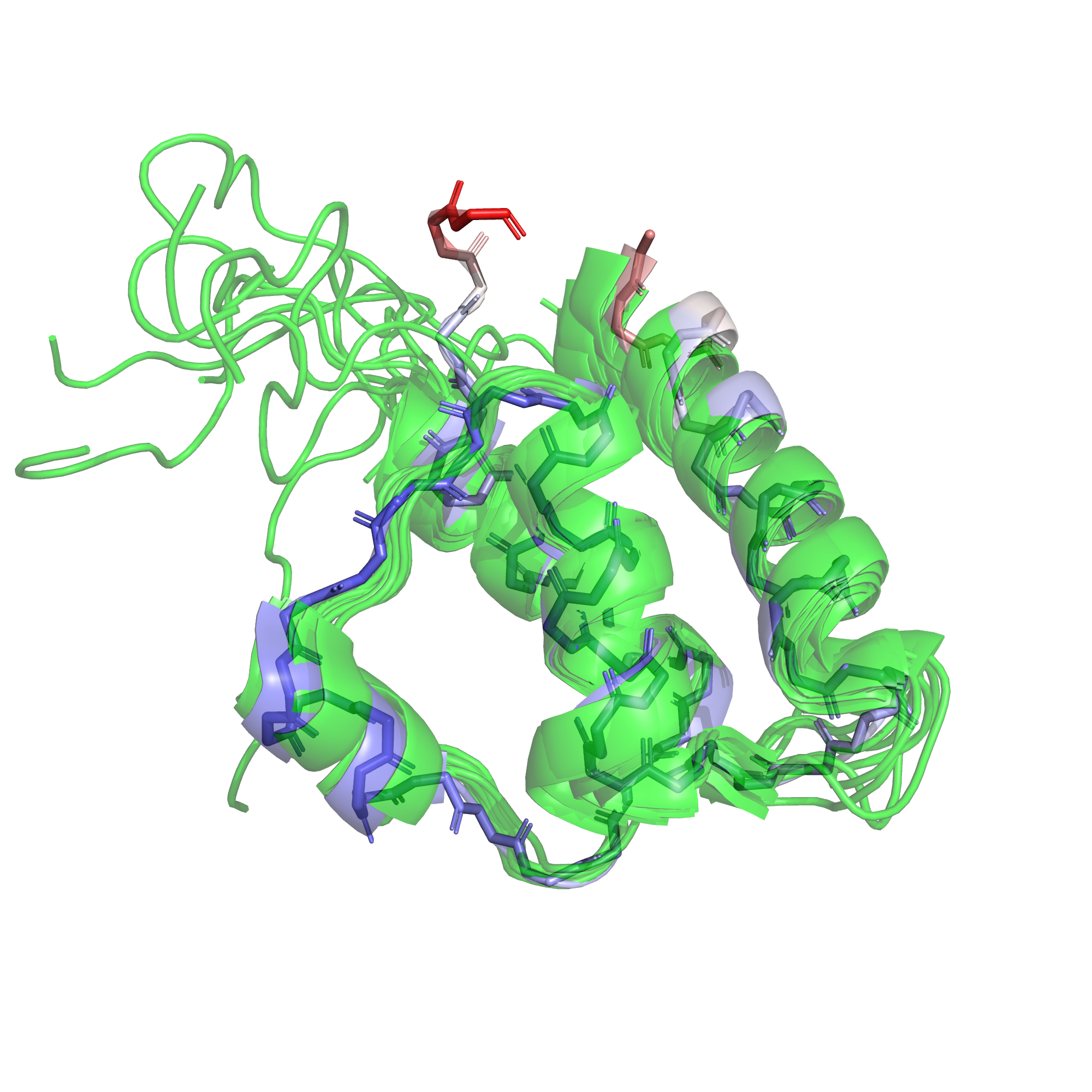
На рис. 4 мы видим те же структуры, но теперь уже без цепи B РСА-структуры и с одним только конформером ЯМР. Кроме того, замечаем, что в ЯМР есть участок из 7 аминокислот на C-конце, отстутствующий в РСА.
Обратив внимание на отдельные аминокислотные остатки (рис. 5) мы замечаем, что в ЯМР-структуре присутствуют атомы водорода, которых нет в РСА. Это неудивительно, поскольку именно химический сдвиг ядер водородов - основной сигнал, измеряемый в ЯМР. Кроме того, на С-конце в целом значительно различается положение аминокислотных остатков в ЯМР и РСА.
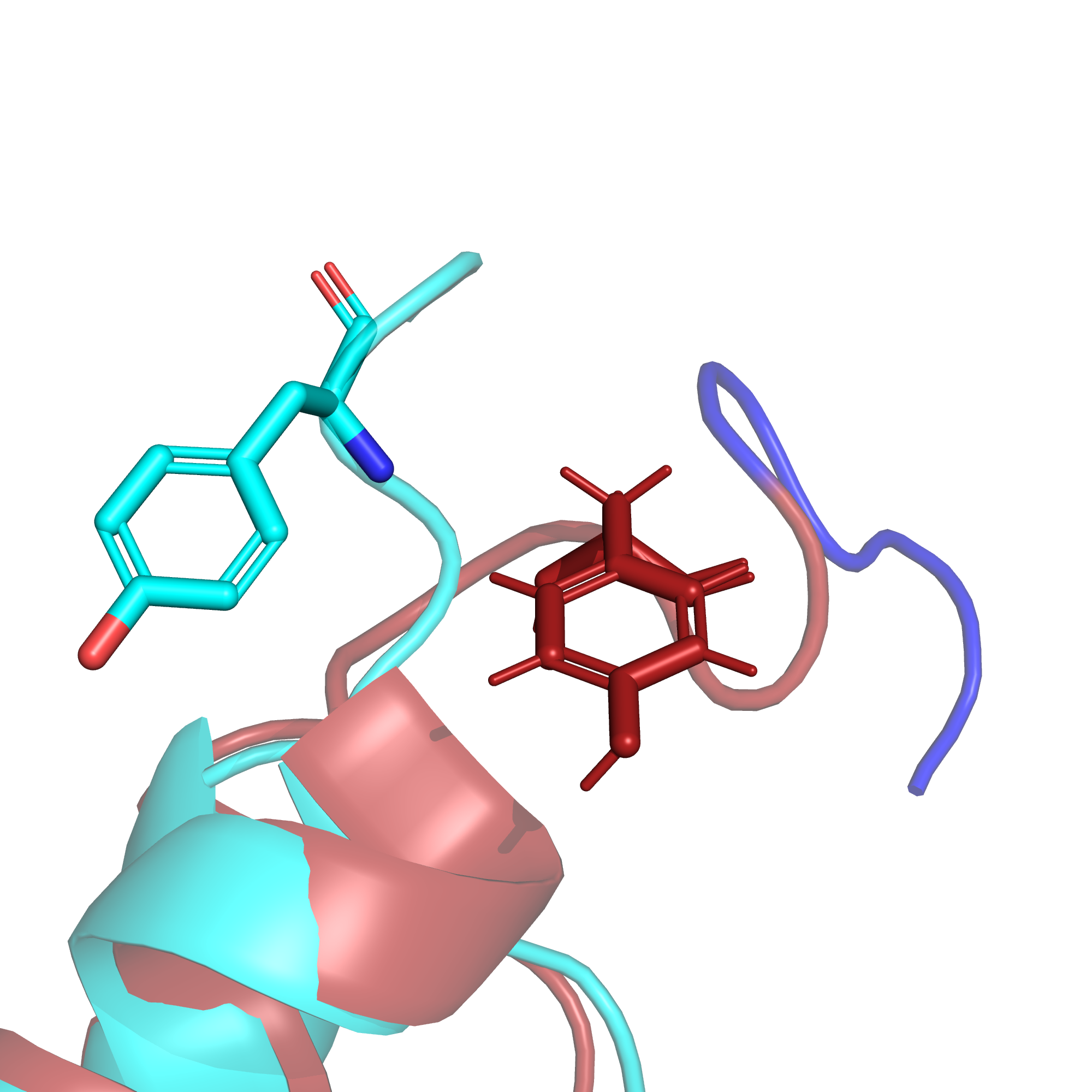
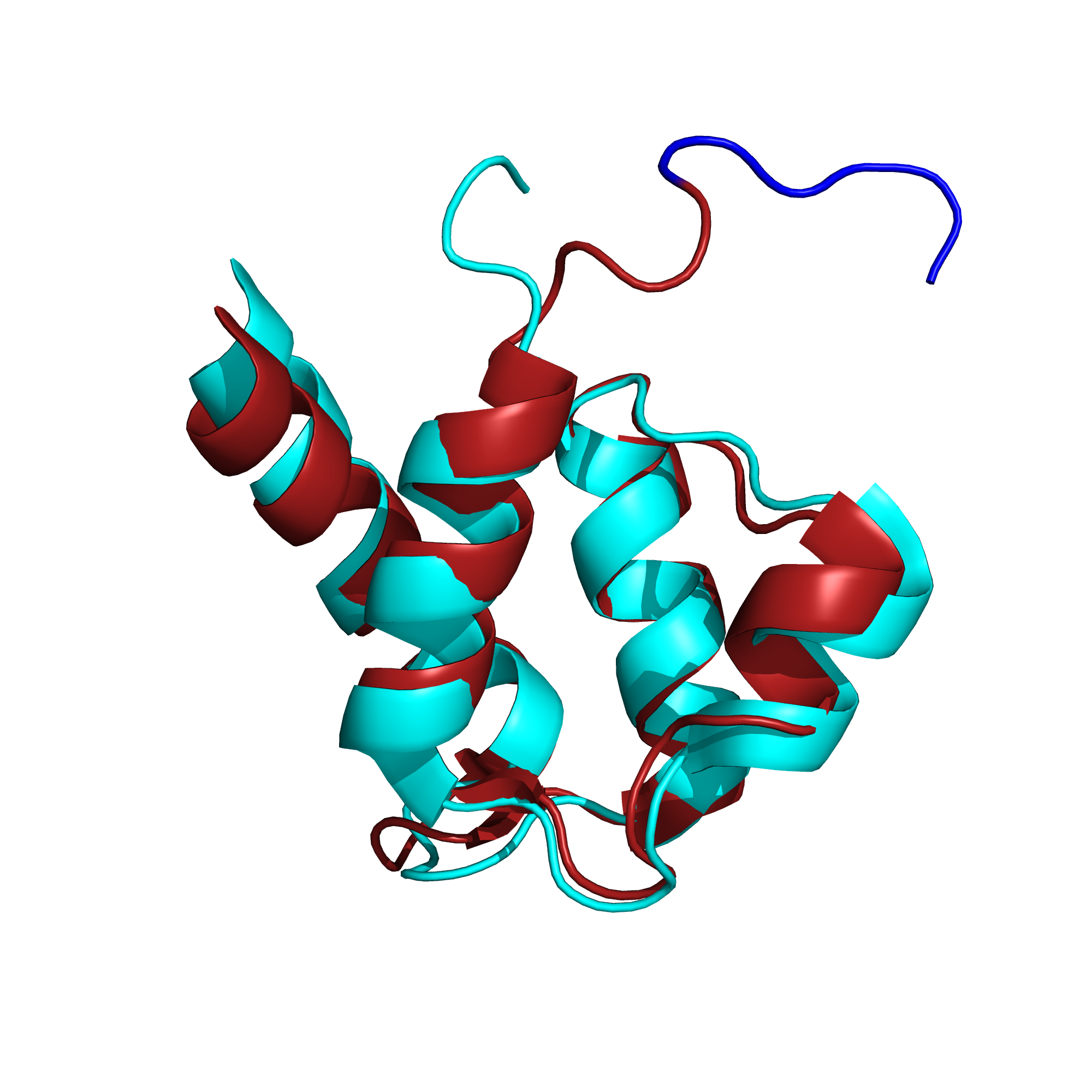
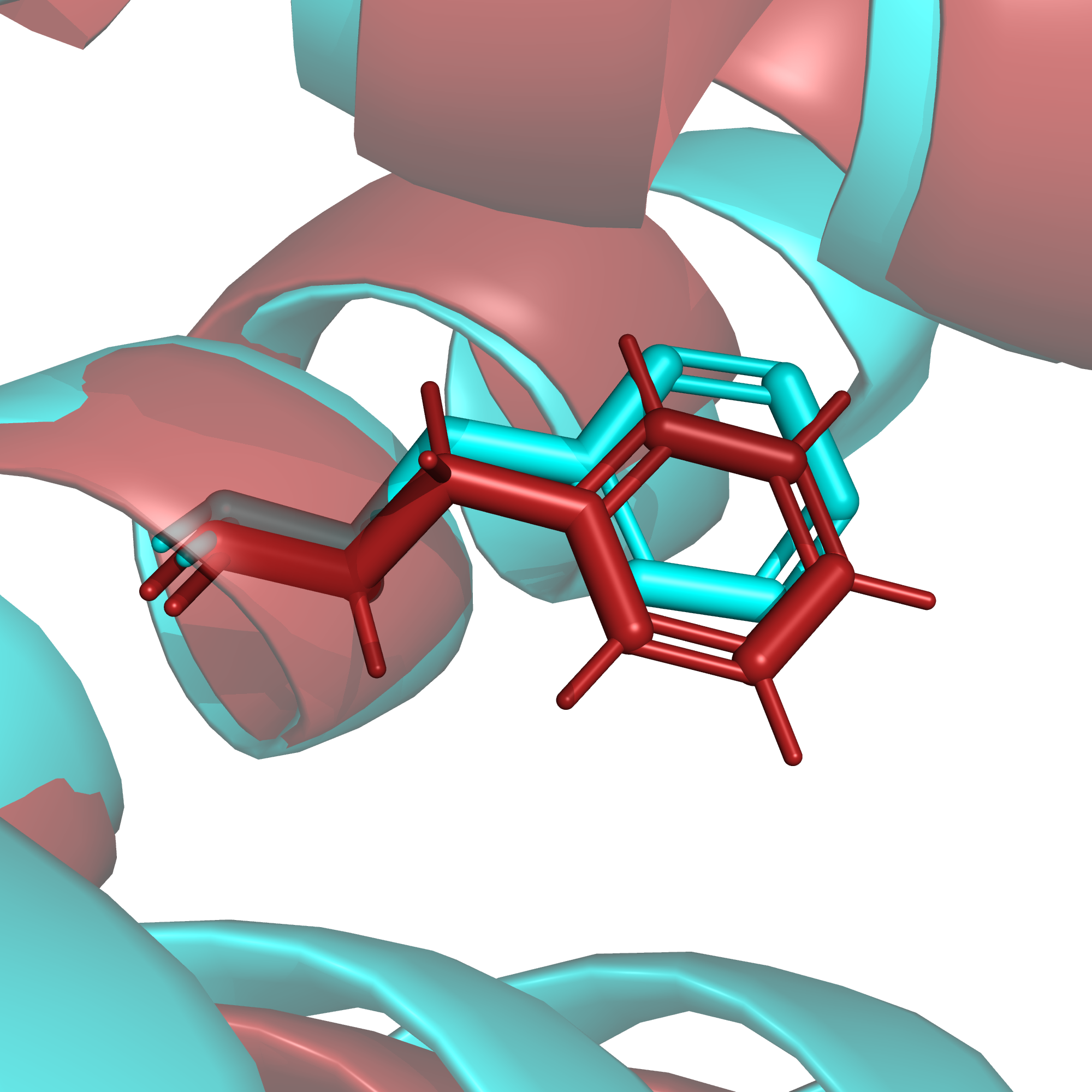
Тем не менее, если мы посмотрим (рис. 6) на аминокислотные остатки внутри альфа-спирали, там расхождение между структурами, полученными разными методами, уже не такое серьезное, хотя небольшой сдвиг все же есть.
Задание 2. RMSF и B-фактор
Различия между конформерами ЯМР необязательно объясняются подвижностью доменов белка, однако в некоторых случаях все-таки могут быть с ней связаны. Для ЯМР-структуры мы можем вычислить RMSF остатков - меру их "подвижности". Сравнив ее со соответствующими средними B-факторами из РСА-структуры, мы сможем приблизительно оценить, насколько разнообразие конформеров связано с подвижностью.
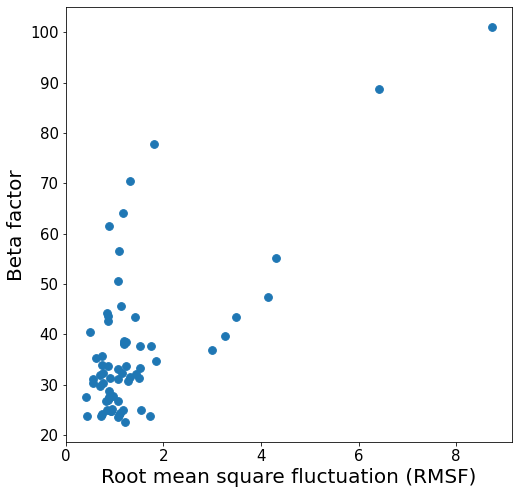
На рис. 7 изображена зависимость B-фактора от RMSF для данных структур. Однозначную зависимость установить нельзя, однако для двух остатков с наиболее высоким B-фактором RMSF также принимает наибольшие значения, а пять остатков со сравнительно высоким RMSF (приблизительно от 3 до 5) имеют не самый маленький B-фактор (приблизительно от 35 до 55). Зависимость для всех этих семи остатков, чей RMSF выше двух, монотонная и вполне похожа на линейную.
Код, использованный мной для выполнение данного задания:
import prody as prd
import numpy as np
import matplotlib.pyplot as plt
nmr_model = prd.parsePDB('6tj3')
xray_model = prd.parsePDB('6tj4')
RMSFs = np.array([np.mean(prd.calcRMSF(res)) for res in nmr_model.iterResidues()])
mean_betas = np.empty(0)
for resi in xray_model.iterResidues():
if 'A' in resi.getChids():
if 'CA' in resi.getNames():
mean_betas = np.append(mean_betas,np.mean(resi.getBetas()))
RMSFs = RMSFs[0:-6]
assert(RMSFs.shape==mean_betas.shape)
fig,ax = plt.subplots(figsize=(8,8))
ax.scatter(RMSFs,mean_betas,s = 60)
ax.set_xlabel("Root mean square fluctuation (RMSF)",fontsize=20)
ax.set_ylabel("Beta factor",fontsize=20)
ax.tick_params(axis='both', which='major', labelsize=15)
fig.show()
Задание 3. Водородные связи в ЯМР и РСА
Также можно сравнить ЯМР и РСА структуры по тому, как различаются в них водородные связи.
| Положение связи | Расстояние в РСА, Å | Процент моделей ЯМР с данной связью | Минимальное расстояние в ЯМР, Å | Максимальное расстояние в ЯМР, Å | Медианное расстояние в ЯМР, Å |
|---|---|---|---|---|---|
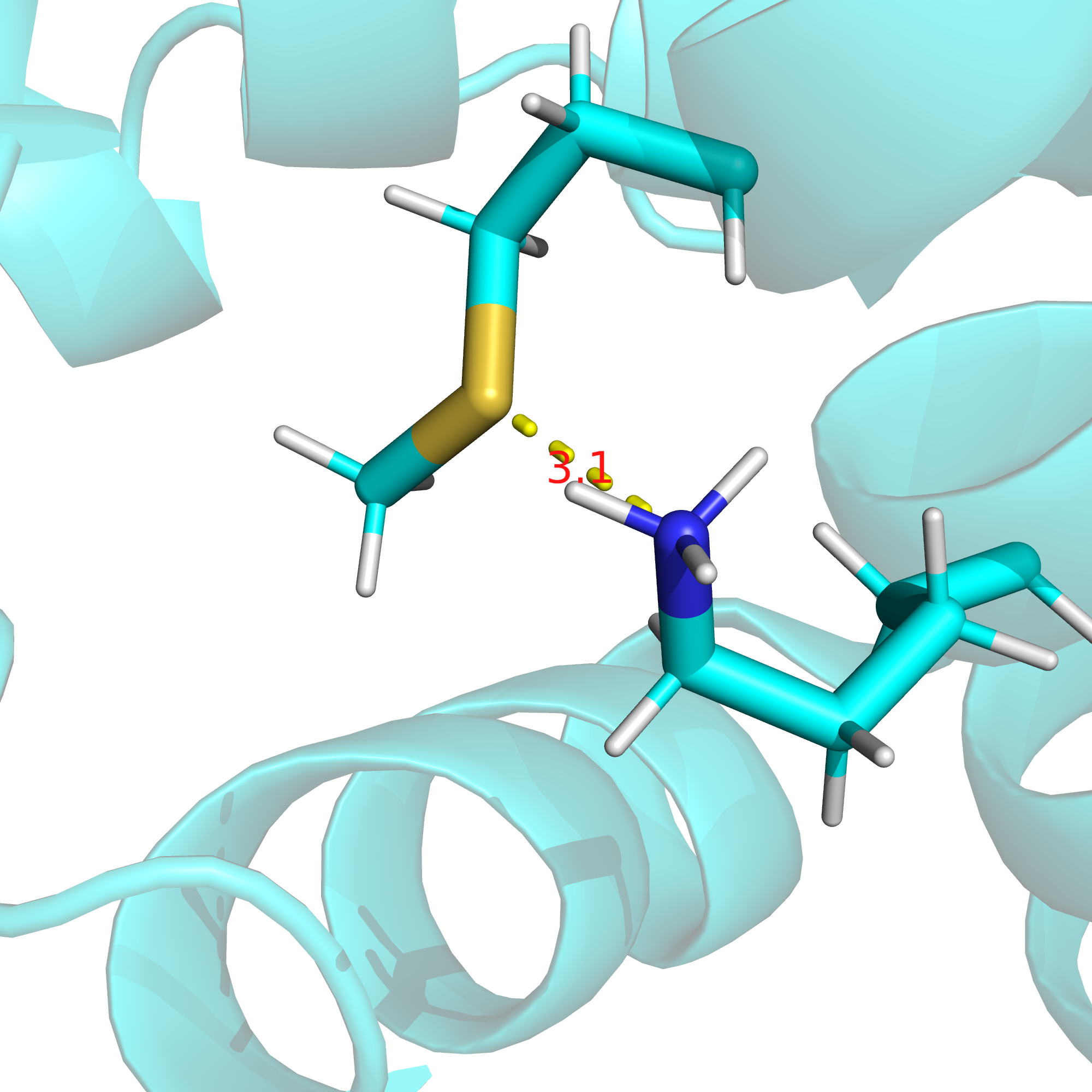
| Остов, альфа-спираль (аспарагиновая кислота-4 и лизин-8). | 3.1 | 100% | 2.97 | 3.77 | 3.38 |
| Боковые цепи в ядре белка (фенилаланин-59 и гистидин-63; в РСА-структуре водородной связи скорее не наблюдается) | 4.09 | 70% | 3.54 | 6.24 | 3.73 |
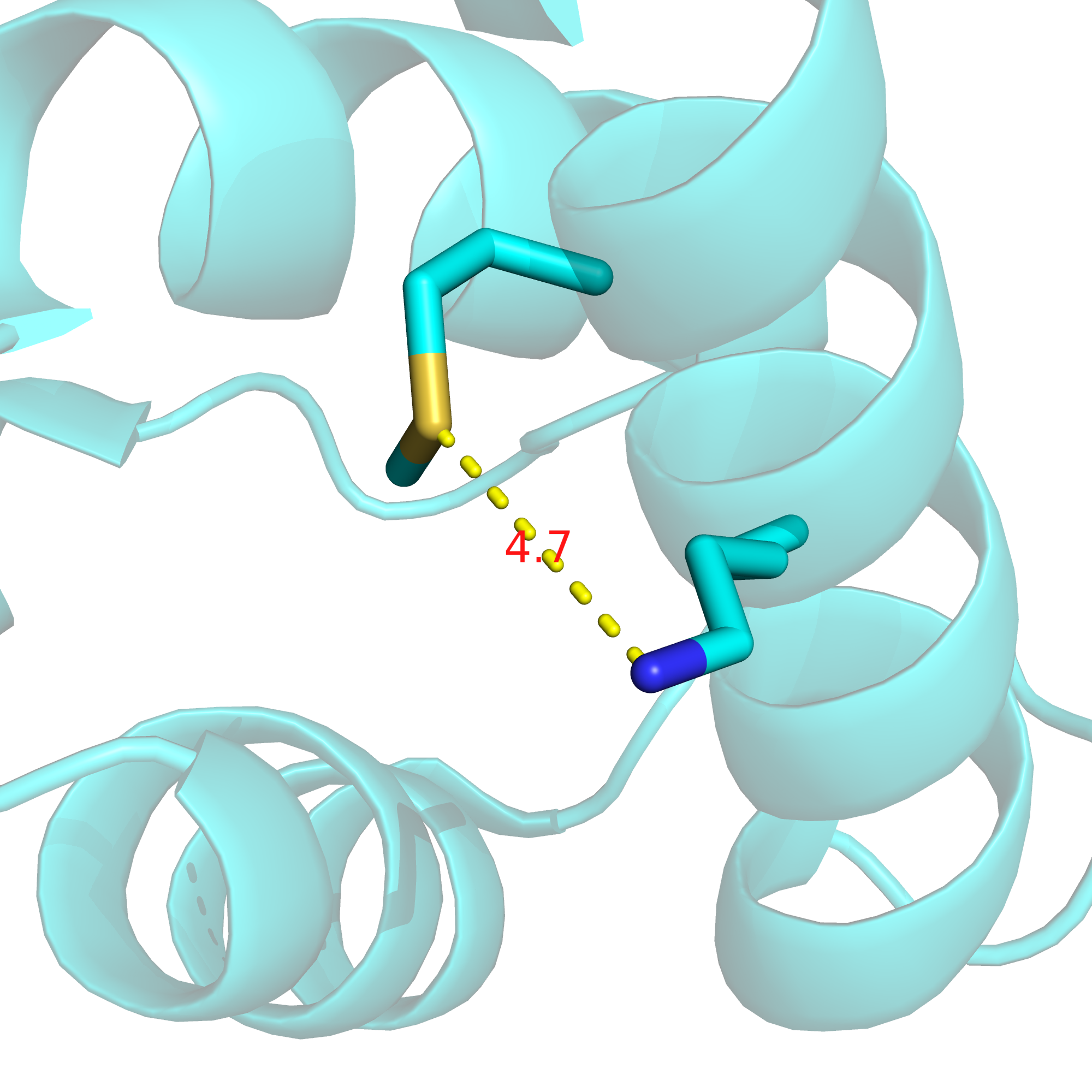
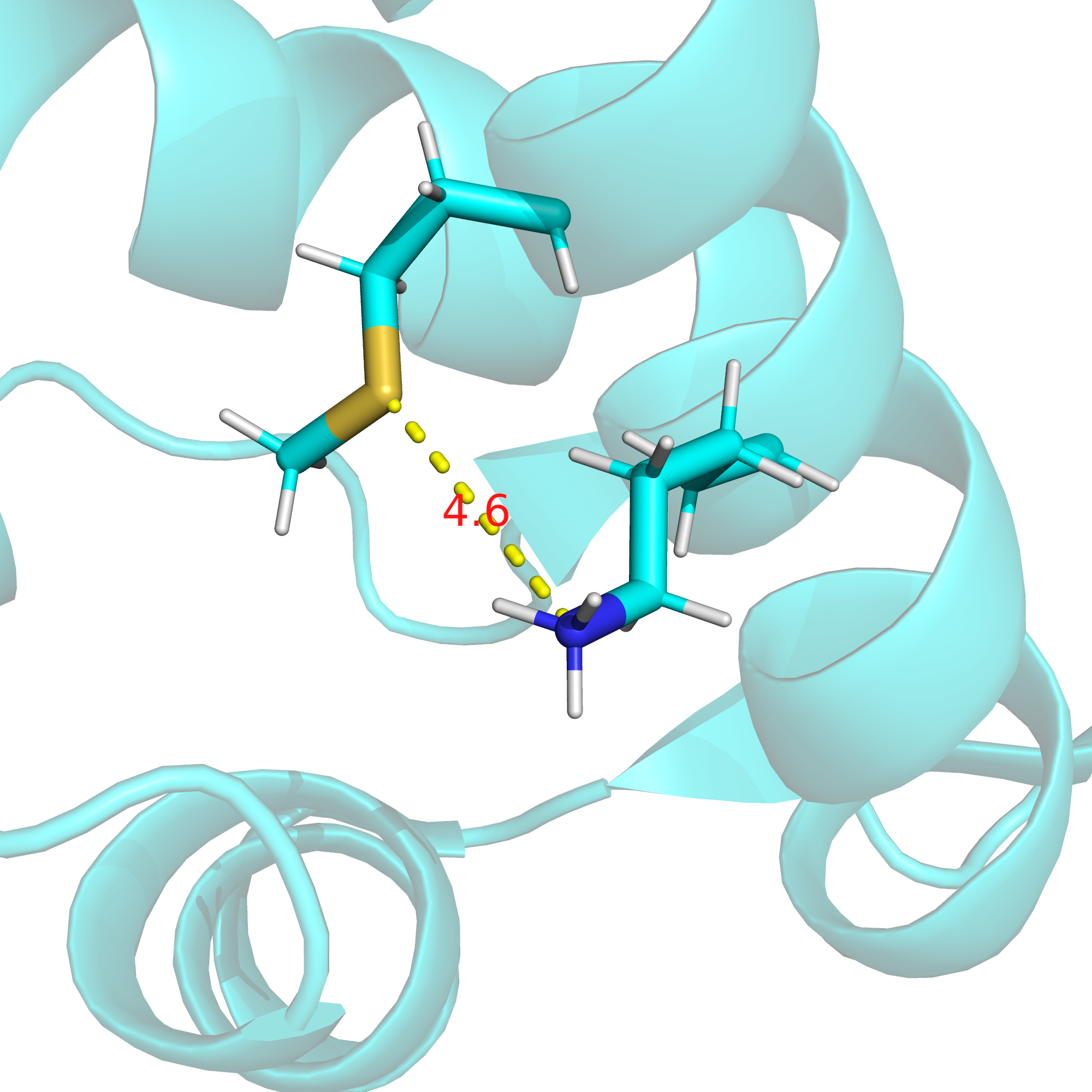
| Боковые цепи на поверхности белка (метионин-5 и лизин-8; в РСА-структуре водородной связи нет) | 4.7 | 10% | 3.07 | 8.62 | 5.83 |
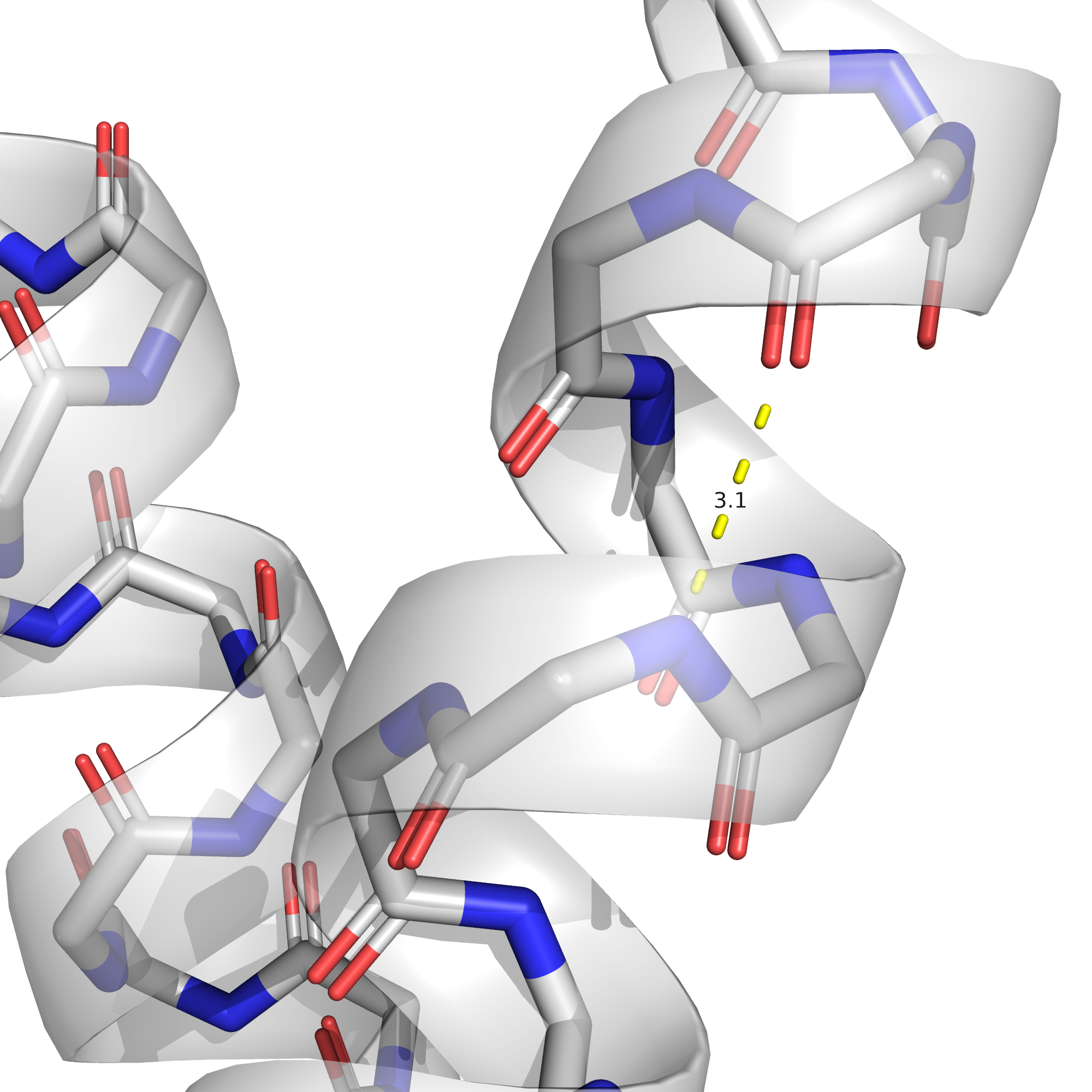
Если взять водородную связь между атомами остова альфа-спирали (рис. 8), она будет наблюдаться во всех конформерах ЯМР, хотя для некоторых из них расстояние между донором и акцептором и является довольно большим (см. Таблицу 1 и рис. 9-10).
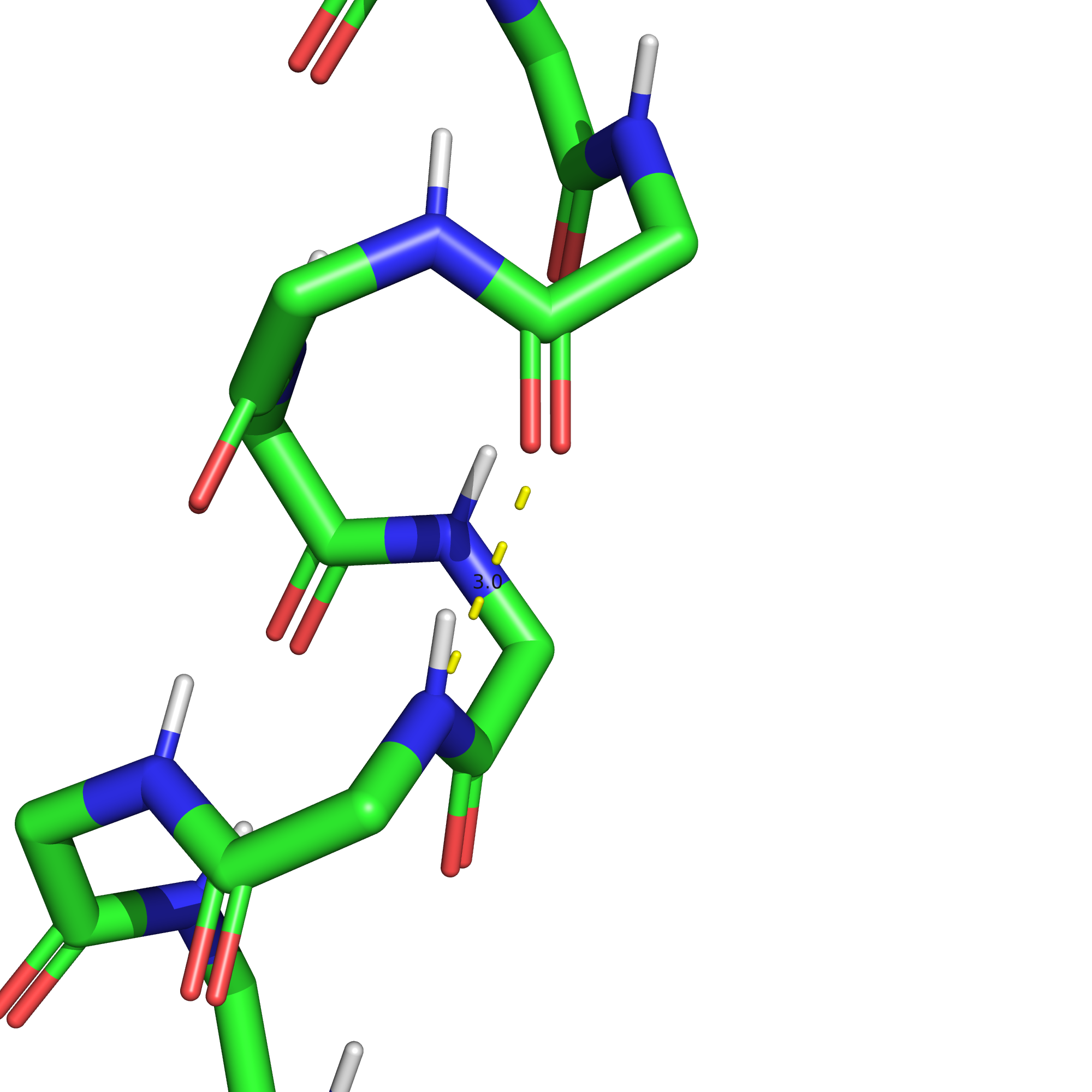
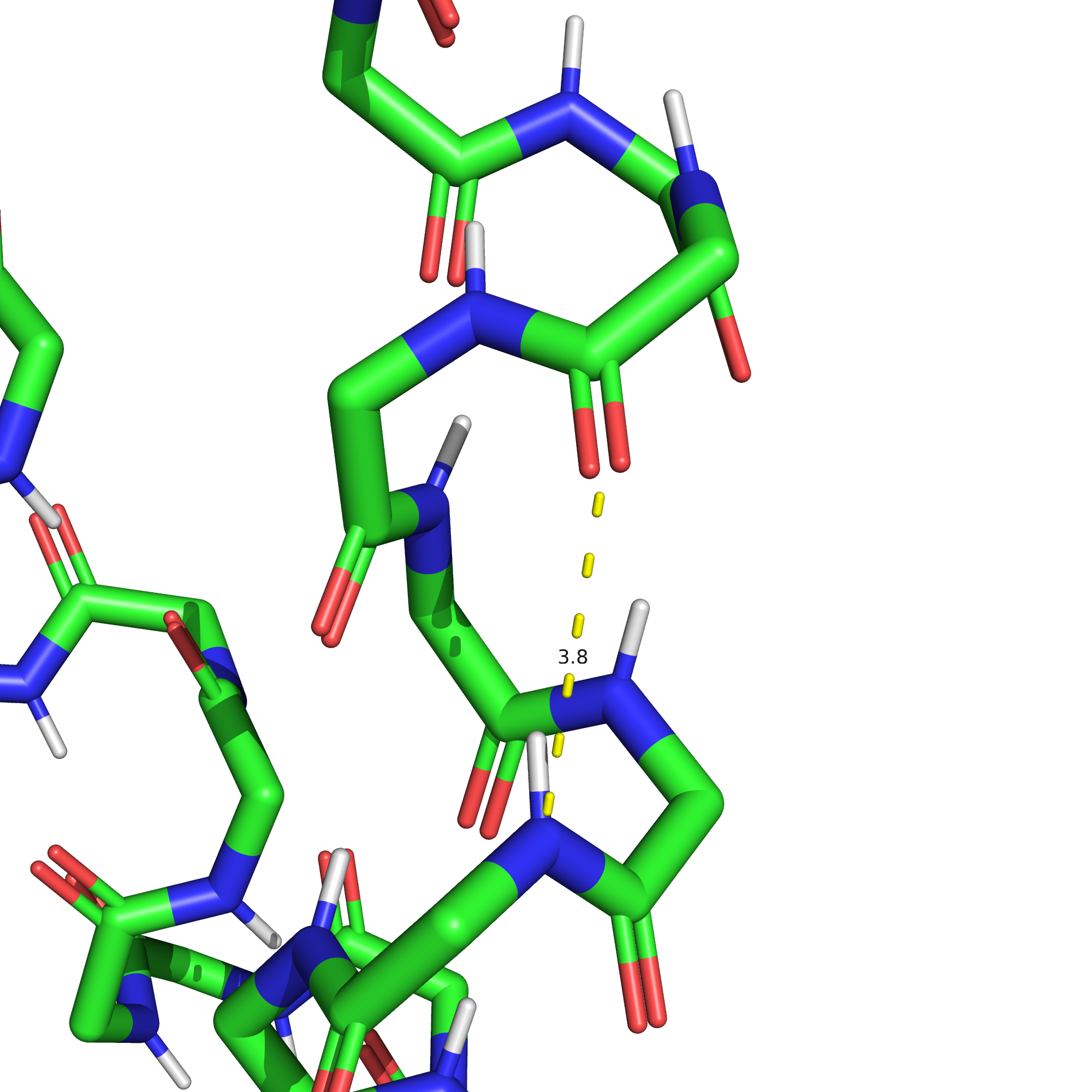
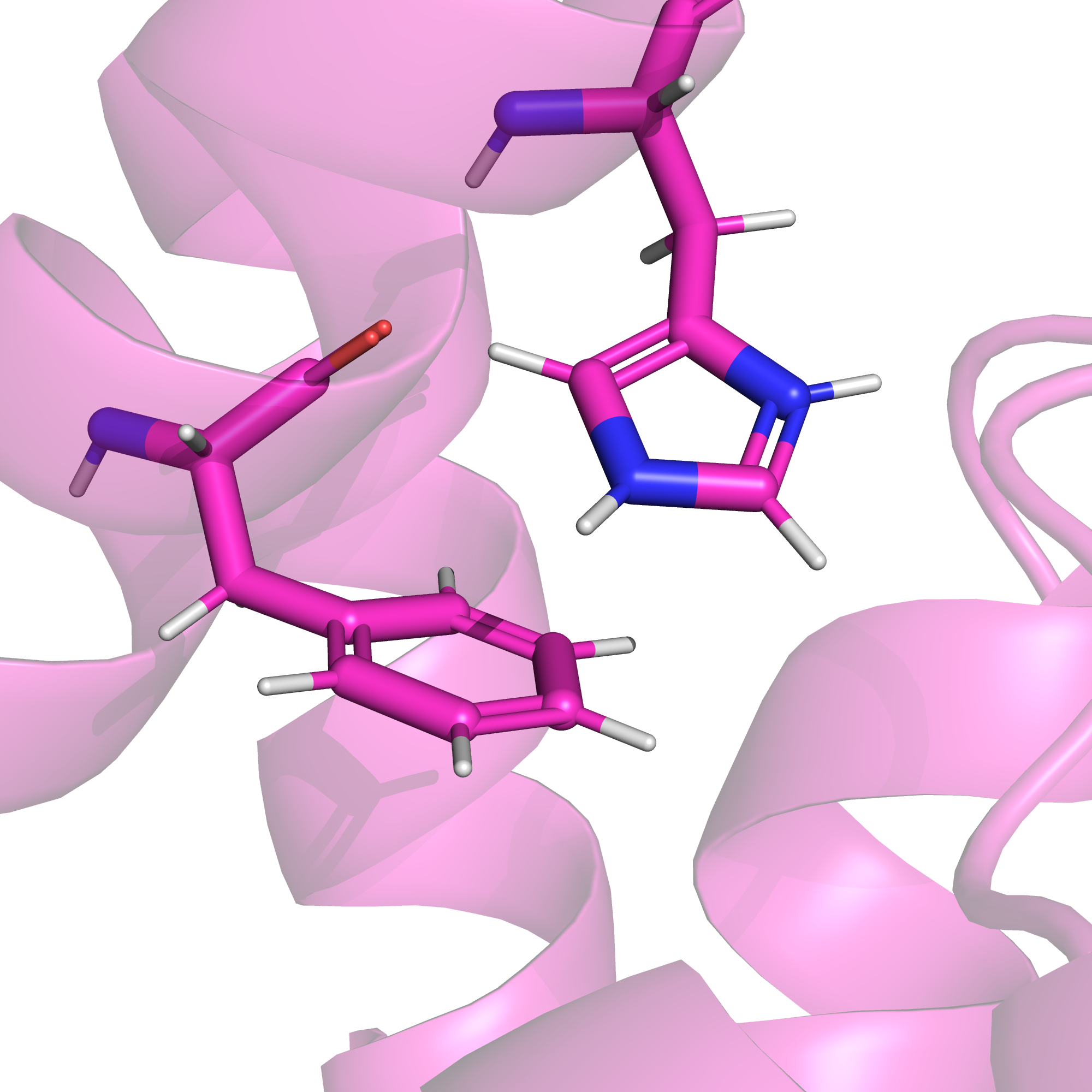
Если же попытаться рассмотреть водородную связь в ядре белка, то мы обнаружим, что в данной структуре боковые цепи, обращенные "внутрь" белка, преимущественно гидрофобные (см. рис. 11). Единственная пара остатков, чьи хотя бы отчасти обращенные внутрь боковые цепи хотя бы в теории могутт образовывать водородную связь в данном белке - это фенилаланин-59 и гистидин-63. Протонированный положительно заряженный гистидин может быть донором, а ароматическое кольцо фенилаланина с повышенной электронной плотностью над и под кольцом - акцептором водородной связи.
Данные остатки в РСА-структуре приведены на рис. 12, информация о расстоянии между ними - в таблице 1. Конечно, нельзя сказать уверенно, что данная связь вообще имеется, но рассчитывать на какое-то взаимодействие в растворе все-таки можно.
Расстояния, приведенные в таблице 1, и для РСА, и для ЯМР посчитаны с помощью Prody. Это расстояние между NE2-азотом гистидина и геометрическим центром кольца фенилаланина. Код, использованный мною, приведен ниже.
На рис. 13 изображены данные остатки в РСА-структуре и во всех конформерах ЯМР-структуры. Видим, что есть две основных возможных конформации: либо остатки повернуты друг к другу и между ними гипотетически возможно образование водородной связи (такое наблюдается в семи ЯМР-конформерах, см. рис. 14), либо остатки совсем отвернуты друг от друга, и даже гипотетически водородной связи между ними быть не может (такая ситуация в остальных трех конформерах, см. рис. 15).
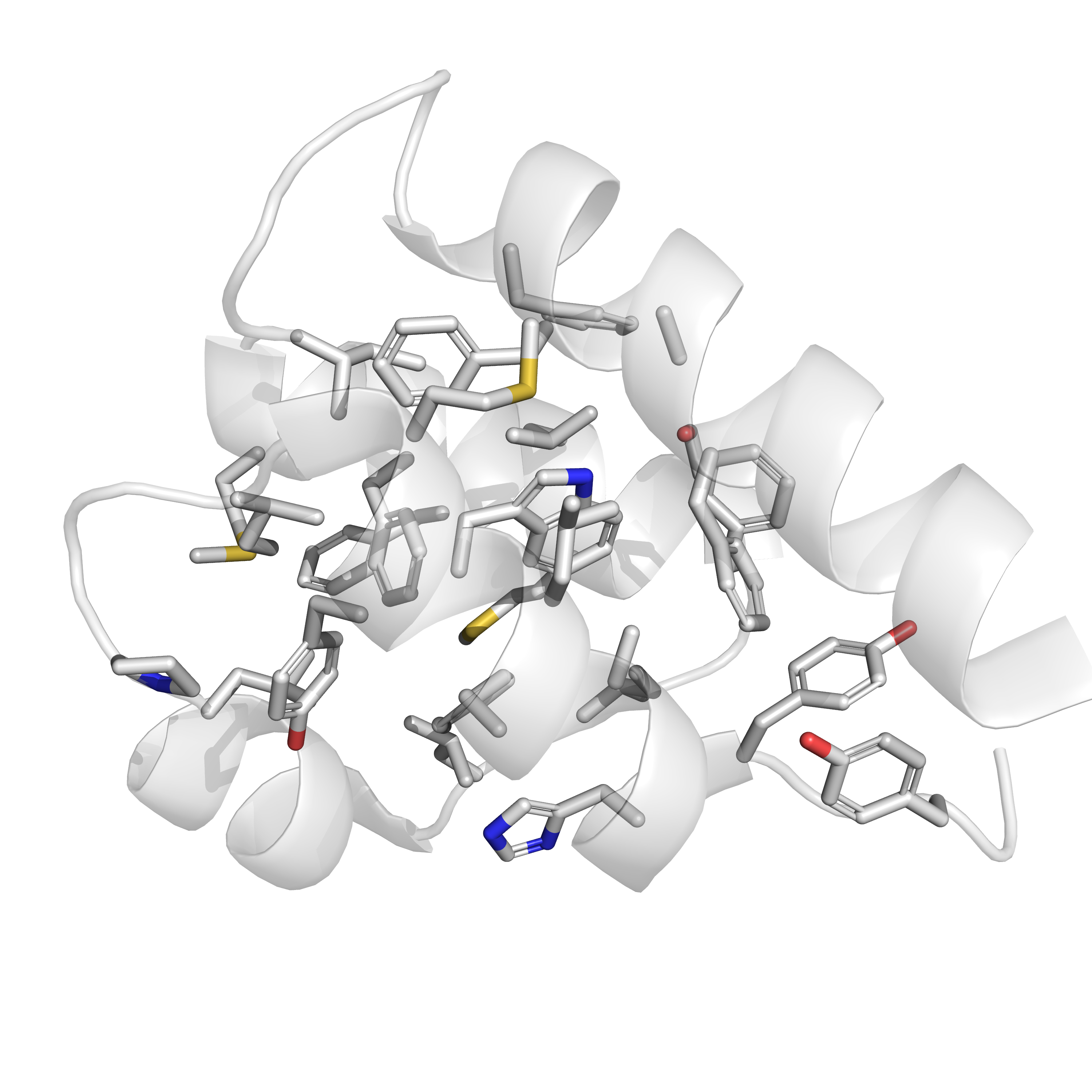
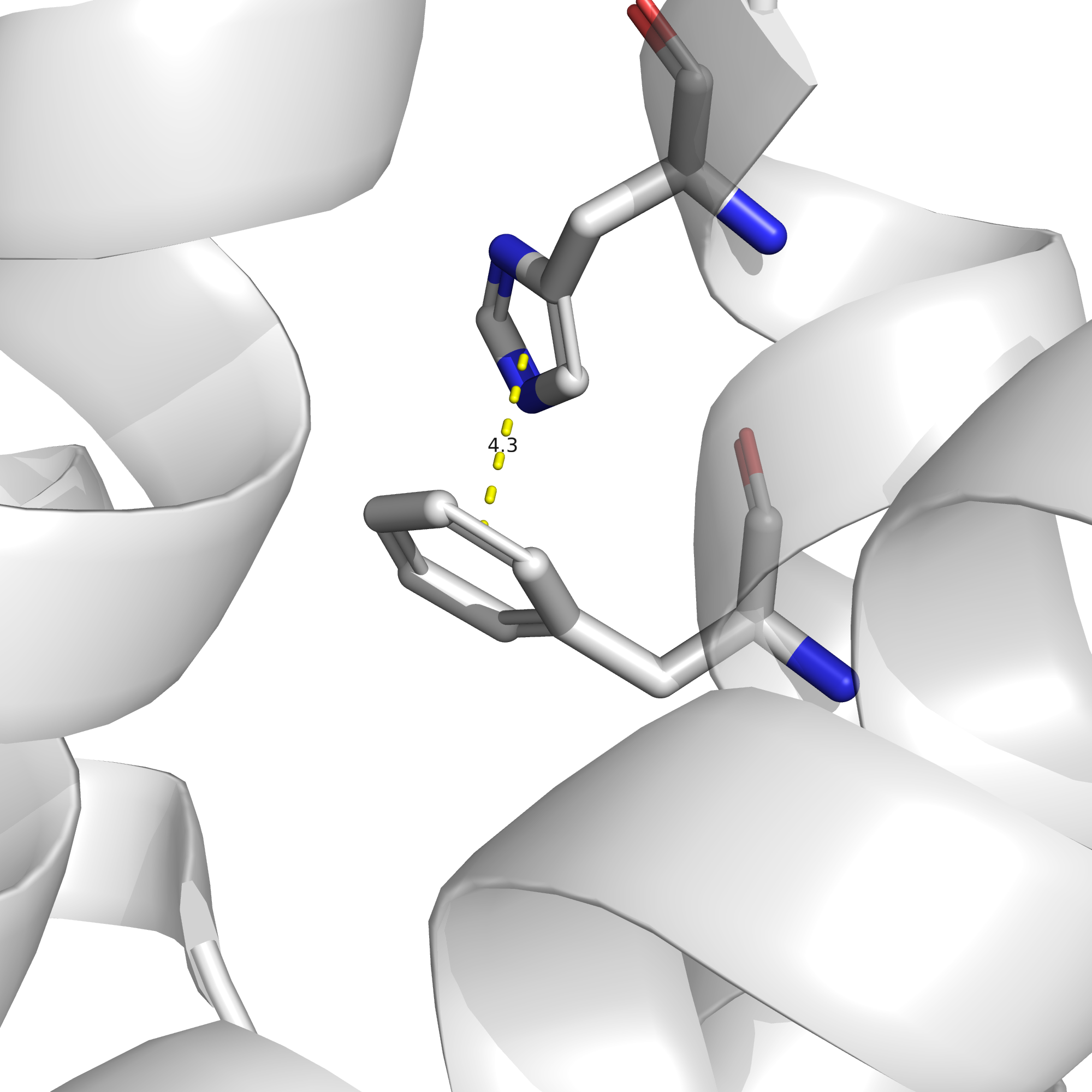
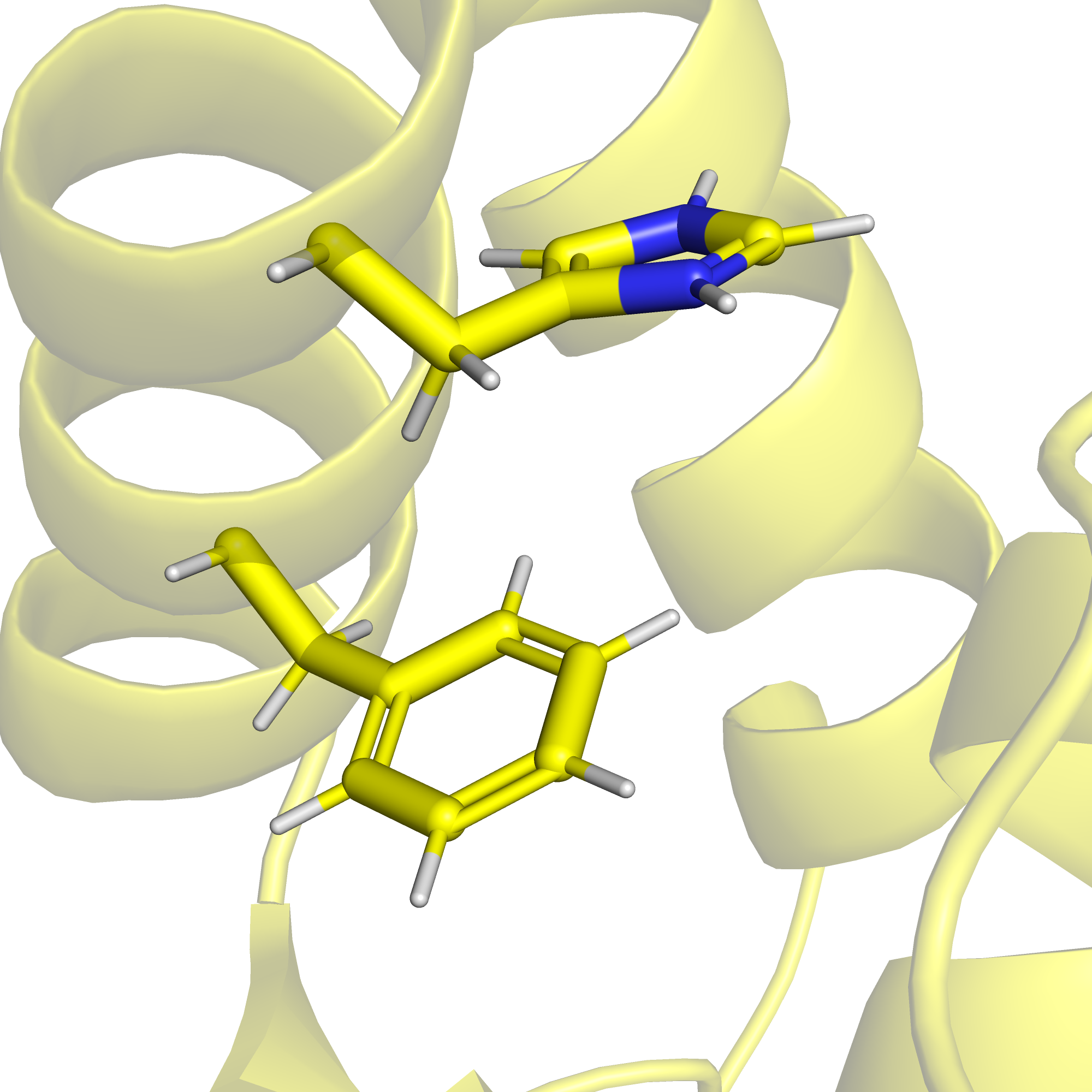
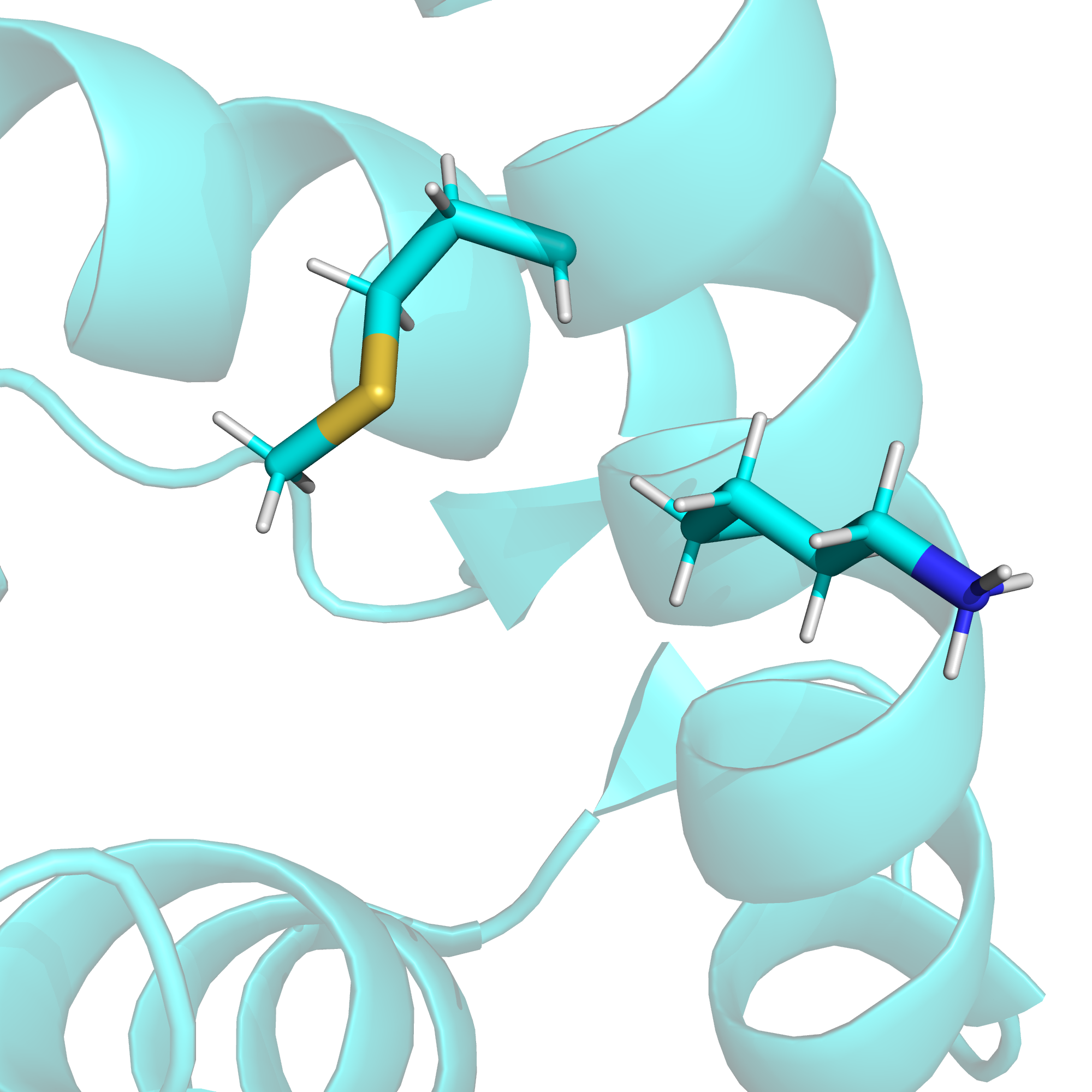
В то же время, на поверхности этого белка аминокислоты преимущественно заряженные: либо отрицательно, либо положительно (см. рис. 16), и мне не удалось найти два таких остатка, между которыми именно водородная связь хорошо бы подтверждалась имеющимися данными. Пришлось отстановиться на связи, которой нет в РСА-структуре и в девяти из десяти ЯМР-конформеров, однако она есть в первом конформере. Это водородная связь между боковыми цепями лизина-8 и метионина-5. На рис. 17 приведено изображение этой связи в первом ЯМР-конформере, на рис. 18 - расстояние между серой и азотом в РСА-структуре, на рис. 19 - расстояние между данными атомами в четвертом ЯМР-конформере, а на рис. 20 - расстояние между данными атомами в седьмом ЯМР-конформере.
Код, использованный мной для выполнение данного задания:
import prody as prd
import numpy as np
nmr_model = prd.parsePDB('6tj3')
asp4 = nmr_model.select('resnum 4 and name O')
lys8 = nmr_model.select('resnum 8 and name N')
distances_backb = []
for asp, lys in zip(asp4.iterCoordsets(),lys8.iterCoordsets()):
distances_backb.append(prd.calcDistance(asp,lys))
print(max(distances_backb),min(distances_backb),np.median(np.array(distances_backb)))
his63 = nmr_model.select('resnum 63 and name NE2')
phe59 = nmr_model.select('resnum 59 and (name CD1 or name CD2 or name CE1 or name CE2 or name CZ or name CG)')
distances_inside = []
for his, phe in zip(his63.iterCoordsets(),phe59.iterCoordsets()):
phe_center = prd.calcCenter(phe)
distances_inside.append(prd.calcDistance(his,phe_center))
print(max(distances_inside),min(distances_inside),np.median(np.array(distances_inside)))
his63 = xray_model.select('resnum 63 and name NE2 and chain A')
phe59 = prd.calcCenter(xray_model.select('chain A and resnum 59 and (name CD1 or name CD2 or name CE1 or name CE2 or name CZ or name CG)'))
prd.calcDistance(his63,phe59)
met5 = nmr_model.select('resnum 5 and name SD')
lys8 = nmr_model.select('resnum 8 and name NZ')
distances_out = []
for asp, lys in zip(met5.iterCoordsets(),lys8.iterCoordsets()):
distances_out.append(prd.calcDistance(asp,lys))
print(max(distances_out),min(distances_out),np.median(np.array(distances_out)))
Список литературы
1. Pazicky, S., Dhamotharan, K., Kaszuba, K. et al. Structural role of essential light chains in the apicomplexan glideosome. Commun Biol 3, 568 (2020). https://doi.org/10.1038/s42003-020-01283-8.
2. FrÉnal K., Foth B.J., Soldati D. (2008) Myosin Class XIV And Other Myosins In Protists. In: Myosins. Proteins and Cell Regulation, vol 7. Springer, Dordrecht. https://doi.org/10.1007/978-1-4020-6519-4_15.