Задание 1. Первый взгляд на структуру
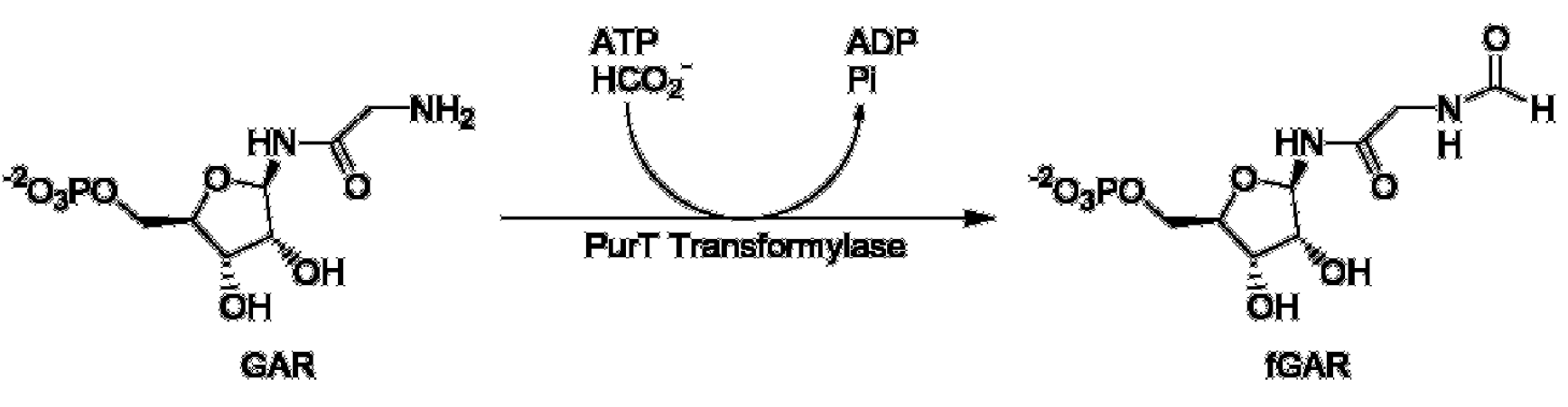
В этом практикуме я работал со структурой 1KJQ. Это структура глицинамидрибонуклеотидтрансформилазы из Escherichia coli, называемой также PurT трансформилазой по названию гена, в комплексе с Mg2+ADP1. Данный фермент играет ключевую роль в синтезе пуринов, катализируя присоединение к глицинамидрибонуклеотиду карбонильной группы формиата с затратой одной молекулы АТФ (см. рис. 1). У человека аналогичная реакция осуществляется белком GART, однако карбонильная группа переносится из 10-формилтетрагидрофолата, а не напрямую из формиата2.
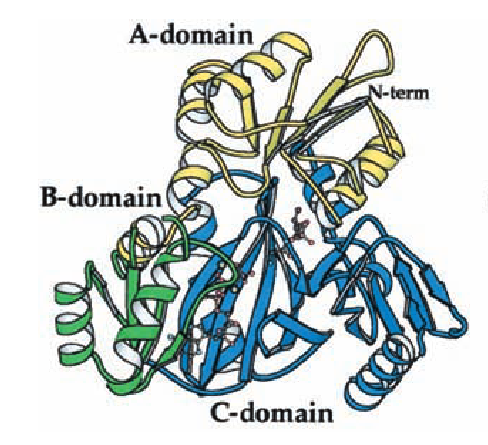
Авторы стуктуры исследовали механизм связывания АТФ в данном ферменте1. Это представляет особый интерес, поскольку глицинамидрибонуклеотидтрансформилаза принадлежит суперсемейству белков ATP-grasp, связывающих АТФ тремя мотивами, называемых A-, B- и C-доменами. Этот механизм сильно менее изучен по сравнению с механизмом связывания через P-петлю, однако также является очень распространенным. Рассматриваемая мной структура 1KJQ - это комплекс фермента с Mg2+ADP, то есть она, в теории, отражает конформацию фермента сразу после гидролиза АТФ.

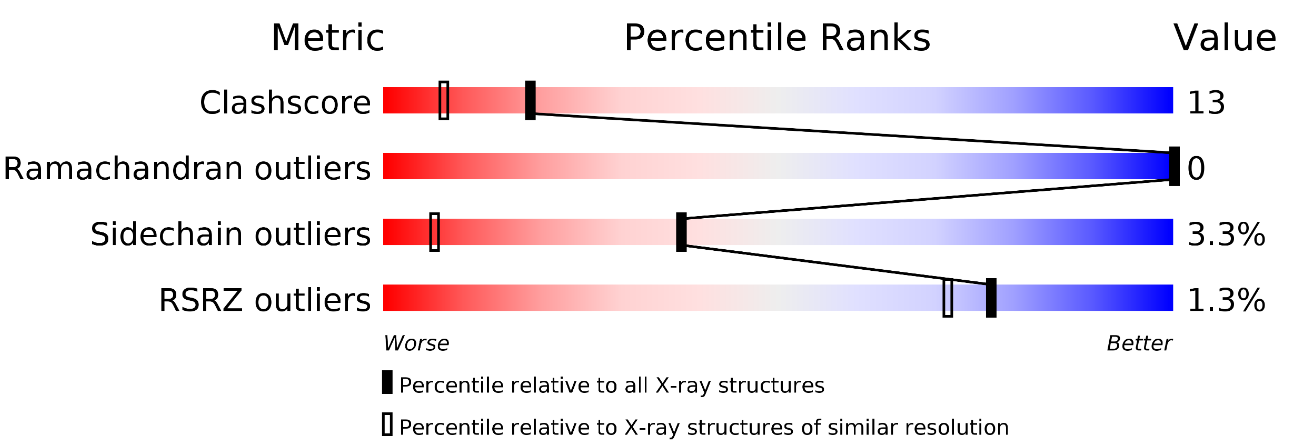
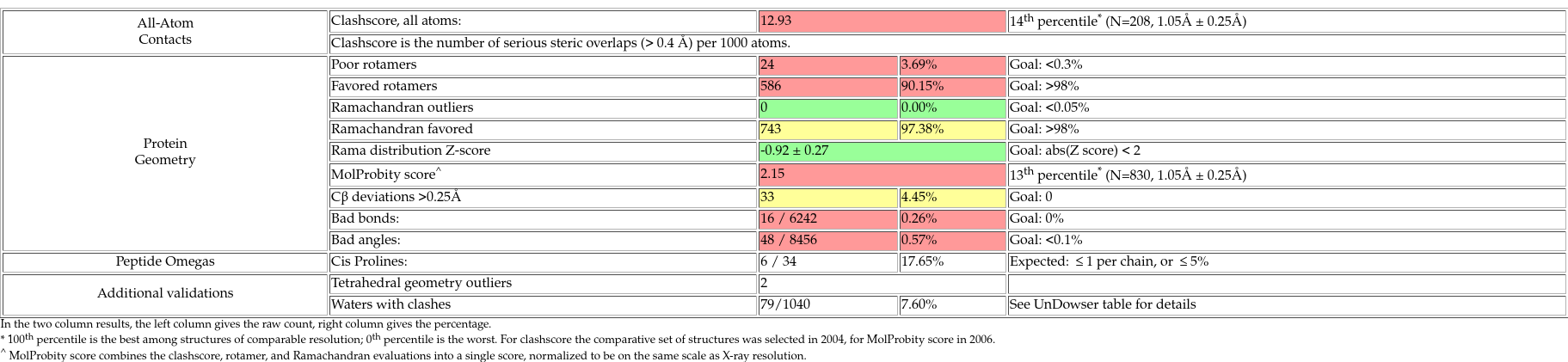
Основные метрики качества данной структуры из отчета в PDB доступны в таблице 1. Размер тестовой выборки при подсчете Rfree - 10%. В целом, метрики качества выглядят неплохо, и Rfree не слишком прешывает R.
При сравнении данной структуры с другими похожего разрешения (рис. 3) сразу замечаем, что у данной структуры есть проблемы с clahscore, то есть многие остатки необычно сближены, а также многие боковые цепи находятся в нестандартной для себя конформации (sidechain outliers). В то же время, не наблюдается аутлаеров по картам Рамачандрана, а также довольно хороший RSR Z-скор (он показывает, насколько хорошо остатки вписаны в электронную плотность, если сравнивать с теми же остатками в других структурах с аналогичным разрешением).
| Метрика | Значение (заявленное авторами) |
|---|---|
| Разрешение | 1.05 Å |
| Полнота данных | 93% |
| R-фактор | 0.188 |
| Rfree-фактор | 0.214 |
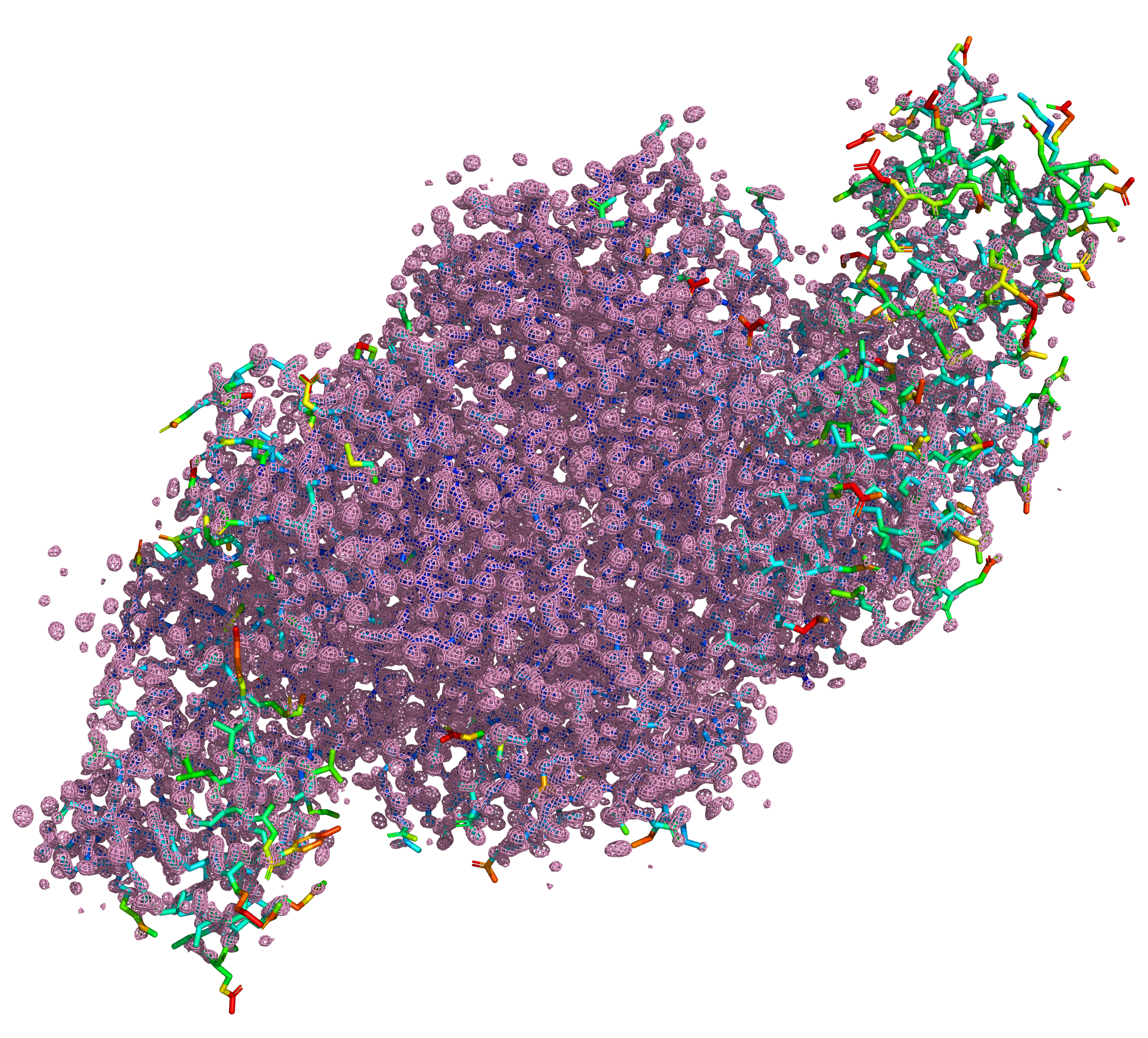
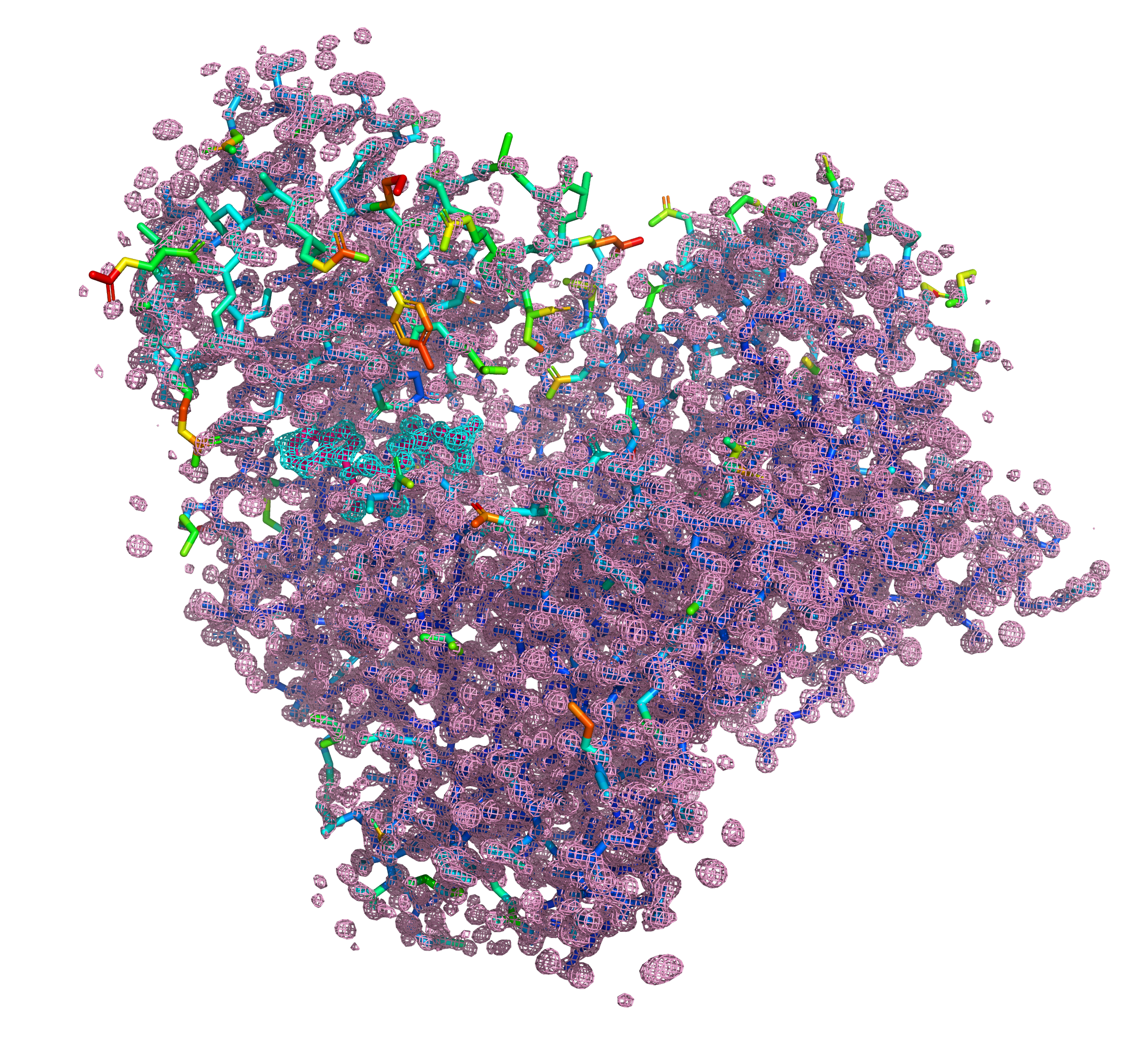
При первом взгляде на стуктуру заметны участки с высоким B-фактором, плохо покрытые сеткой электронной плотности (рис. 4).
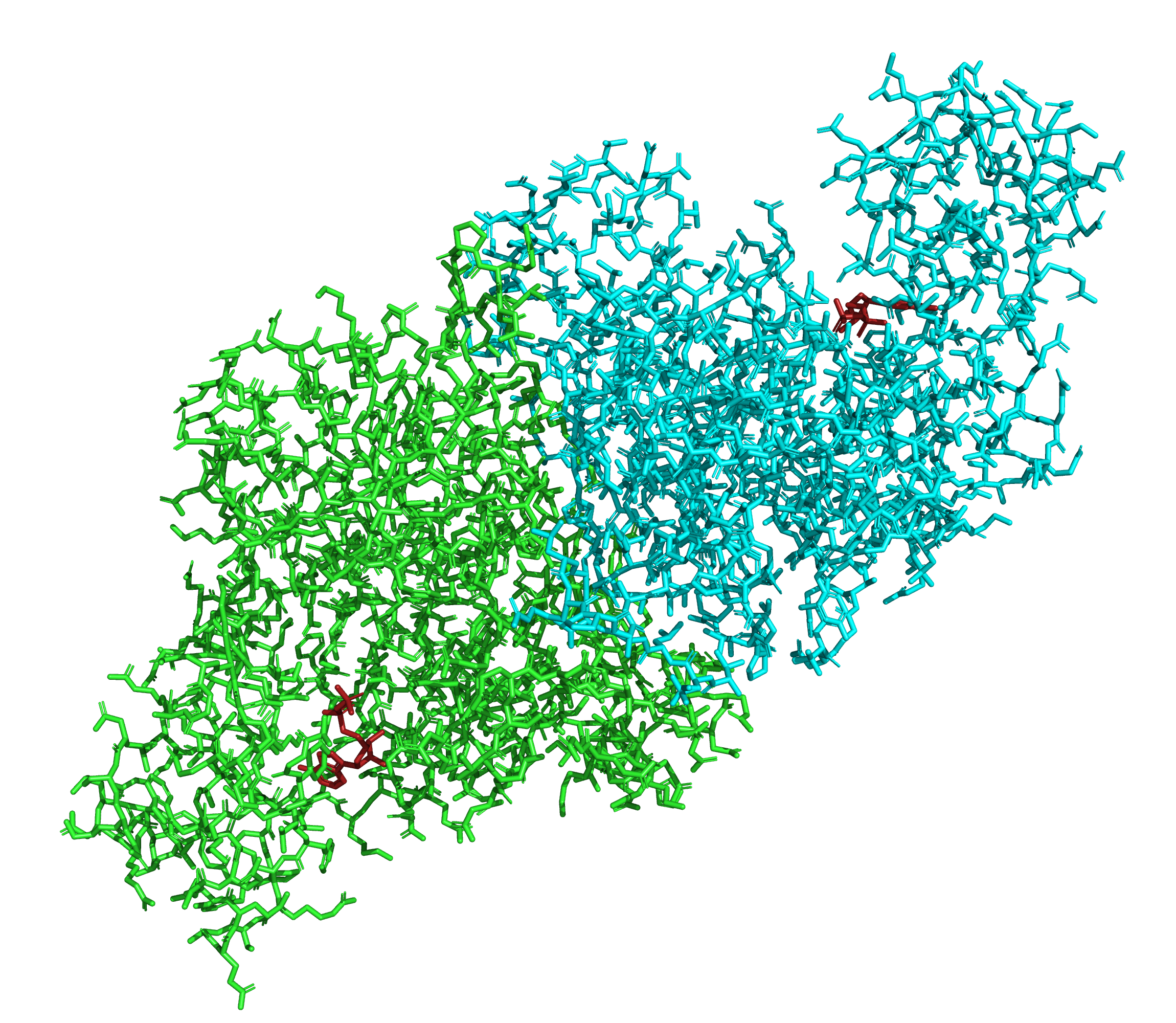
Стоит отметить, что данный белок состоит из двух субъединиц, каждая из которых содержит активный центр (рис. 5). При этом качество структуры субъединицы I выше (и это видно даже из рис. 4), чем субъединицы II, и авторы в своем исследовании рассматривали только первую субъединицу.
Каждая субъединица данного белка состоит из трех доменов (A, B и C; рис. 7), и субстрат связывается с доменом A, а ATP - между доменами B и C1. Если мы прицельно посмотрим на субъединицу I, то увидим, что участки, хуже всего покрытые электронной плотностью, а также обладающие самыми высокими B-факторами, расположены преимущественно в домене B.
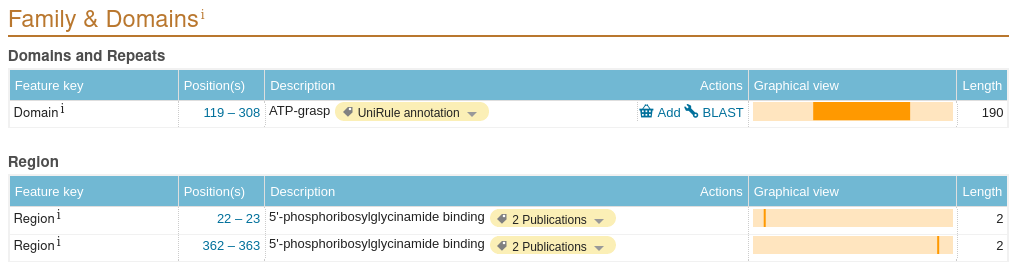
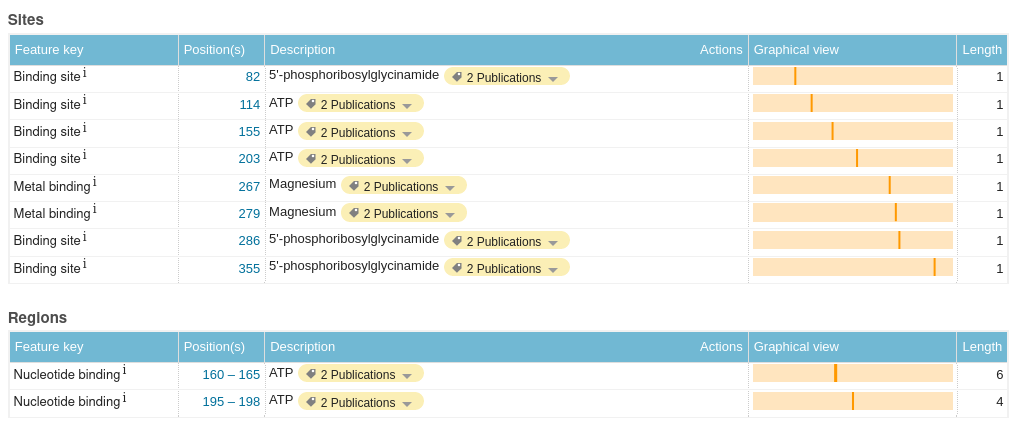
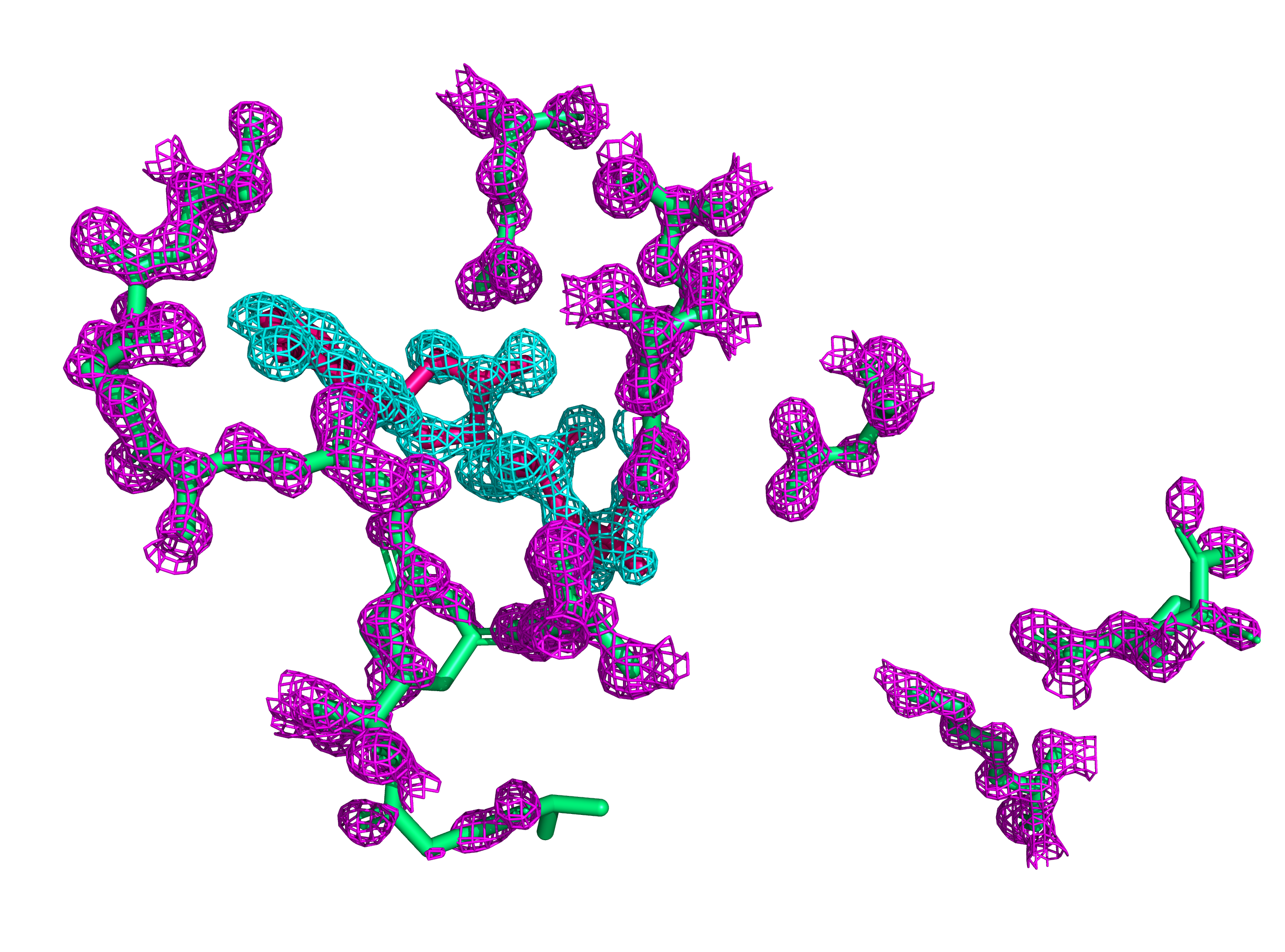
Если мы обратимся к базе данных Uniprot (ID данного белка - P33221), то обнаружим, там информацию о ключевых сайтах и участках, а также доменах и регионах данного белка (рис. 8, 10). При взгляде на качество электронной плотности для данных участков становится понятно, что хотя в целом структура домена ATP-grasp не очень выского качества (рис. 9), ключевые для связывания субстрата и АТФ остатки довольно хорошо вписаны в соответствующую им электронную плотность (рис. 11).
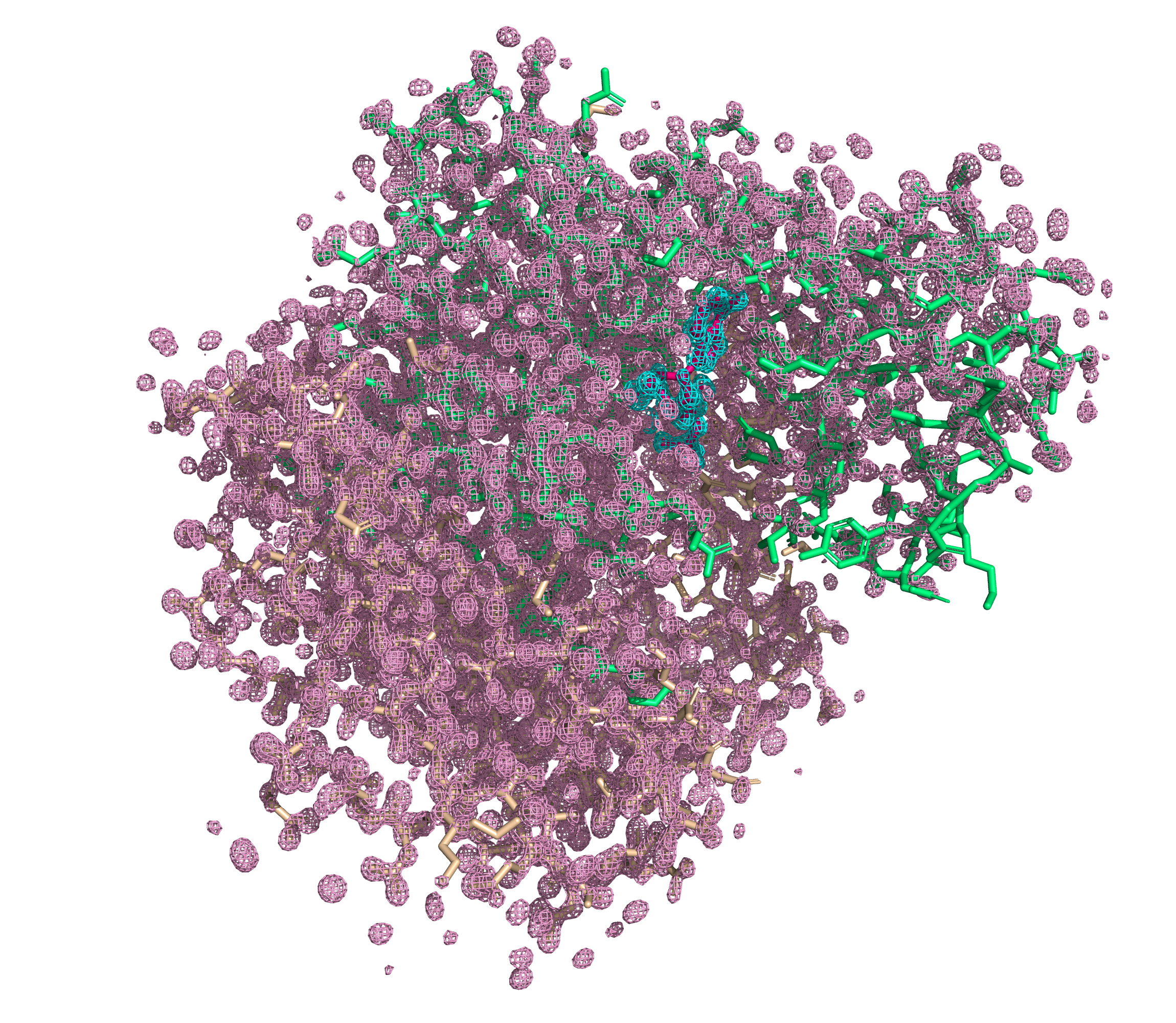
Таким образом, при первом взгляде на струкуру заметно, что не для всех участков можно быть уверенным в её качестве. При этом одна из субъединиц значительно лучше покрыта электронной плотностью. Если ставить своей целью изучение именно механизма реакции и механизма связывания АТФ, то на первый взгляд сложно однозначно оценить, насколько участки с низким качеством мешают исследованию: с одной стороны, в одном из доменов, принимающих участие в связывании АТФ, многие боковые цепи и даже участки остова плохо покрываются электронной плотностью, однако эти проблемы могут при детальном рассмотрении оказаться не слишком серьезными.
Задание 2. Маргинальные остатки
Цель данного задания - анализ структуры в MolProbity и CheckMyMetal для установления, в частности, маргинальных остатков и ионов, а также детальное рассмотрение нескольких из данных остатков.
Результаты анализа данной структуры в MolProbity доступны на рис. 12. Полученная информация о качестве структуры согласуется с приведенной выше краткой инфографикой из PDB, что неудивительно, поскольку PDB, собственно, и использует MolProbity для контроля качества.
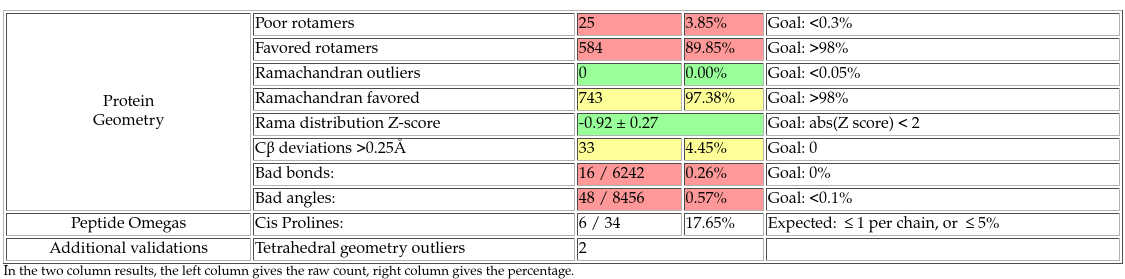
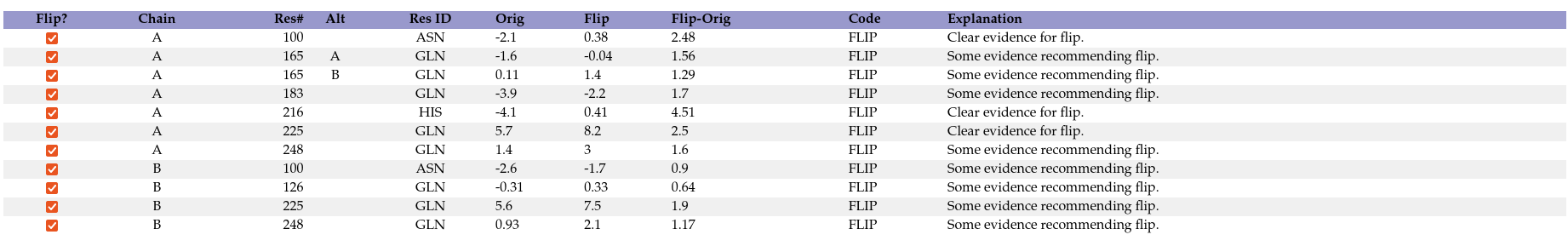
Как уже было сказано, в данной структуре хорошее качество остатков по картам Рамачандрана, однако всё довольно плохо с углами связей и ротамерами остатков и их боковых цепей. Кроме того, есть ряд позиций, требующих разворота после проверки инверсий His/Asn/Gln (рис. 13). Clashscore, как и было сказано выше, также оставляет желать лучшего (структура попадает в 15% самых плохих по этому параметру среди структур аналогичного разрешения).
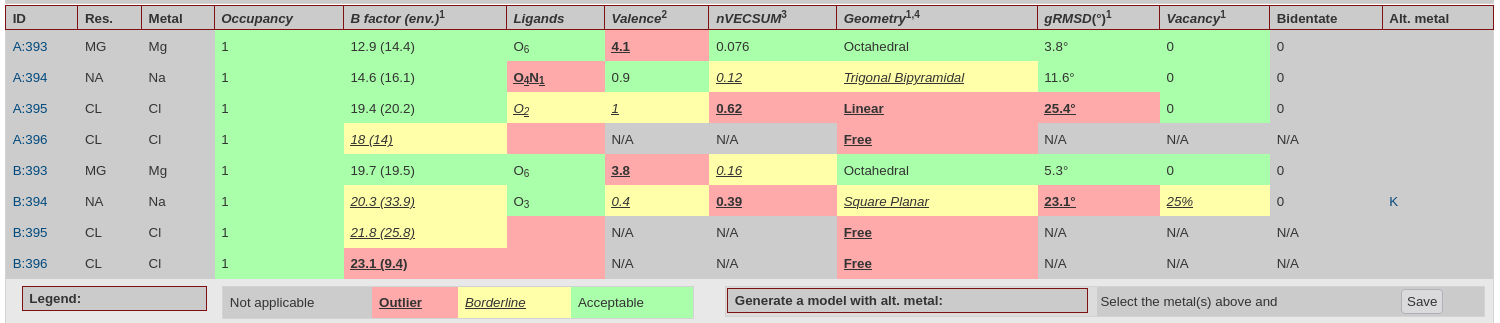
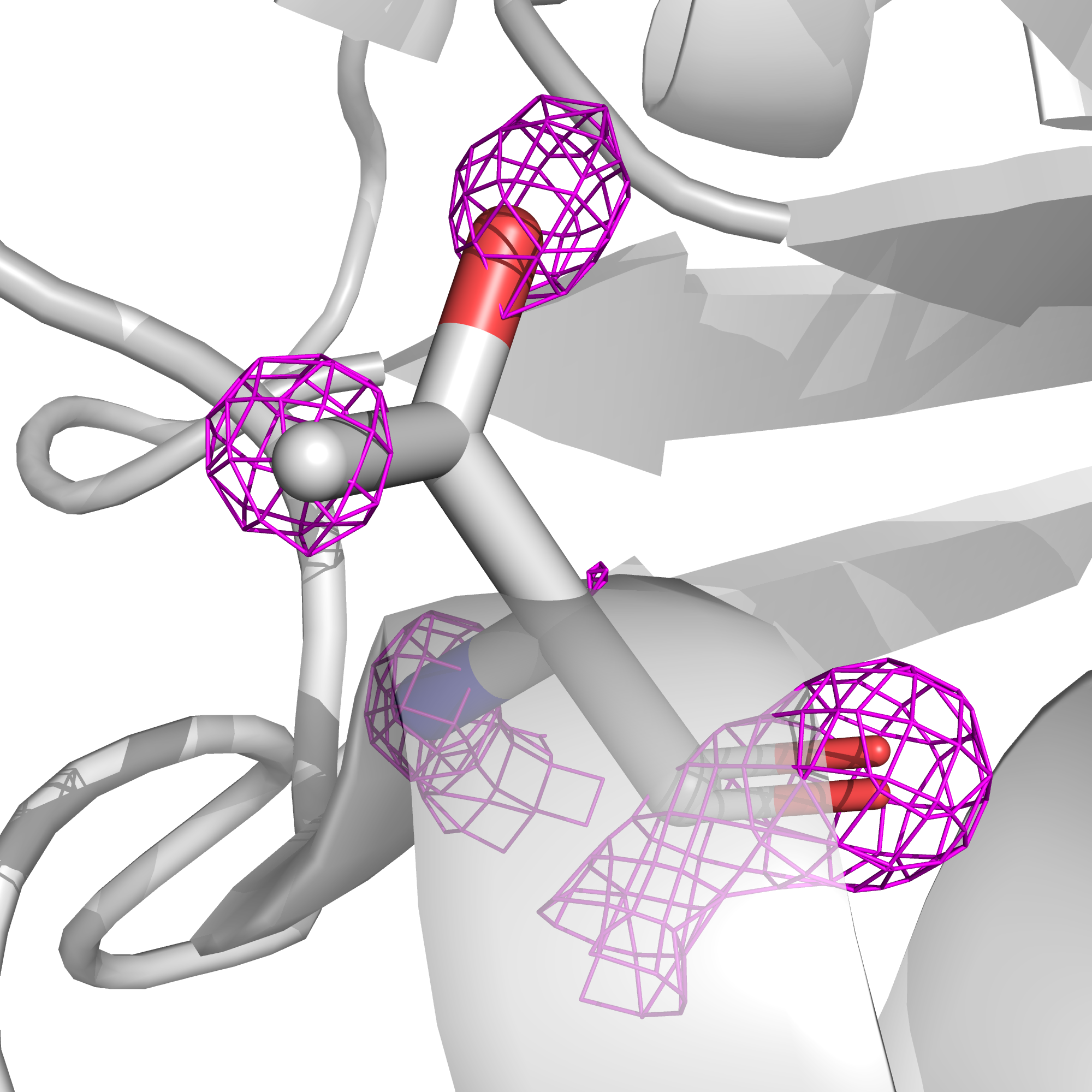
Данная структура содержит ионы металлов, качество положения которых можно оценить с помощью ChekMyMetal (рис. 15). Видим, что в целом качество структуры сайтов ионов для цепи B хуже, чем для цепи A, что подтверждает возникающее еще по электронной плотности предположение, что для исследования лучше использовать именно цепь А (см. задание 1). Стоит сказать, что положение иона Mg представляет значительный интерес, поскольку он играет важную роль в связывании АТФ в данном белке. В цепи A данный ион (первая строка в табличке), по-видимому, вполне разумно расположен: аутлаером он является только по валентности.
Для детального рассмотрения я выбрал 5 маргинальных остатков по данным MolProbity Multi-criterion chart.
1. Остаток аспарагиновой кислоты-229 цепи А.
2. Остаток треонина-88 цепи А.
3. Остаток лейцина-99 цепи А.
4. Остаток фенилаланина-307 цепи А.
5. Остаток аргинина-191 цепи B.




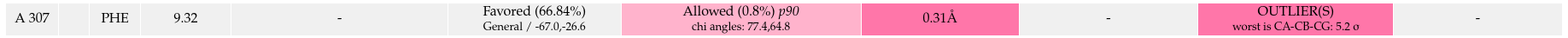

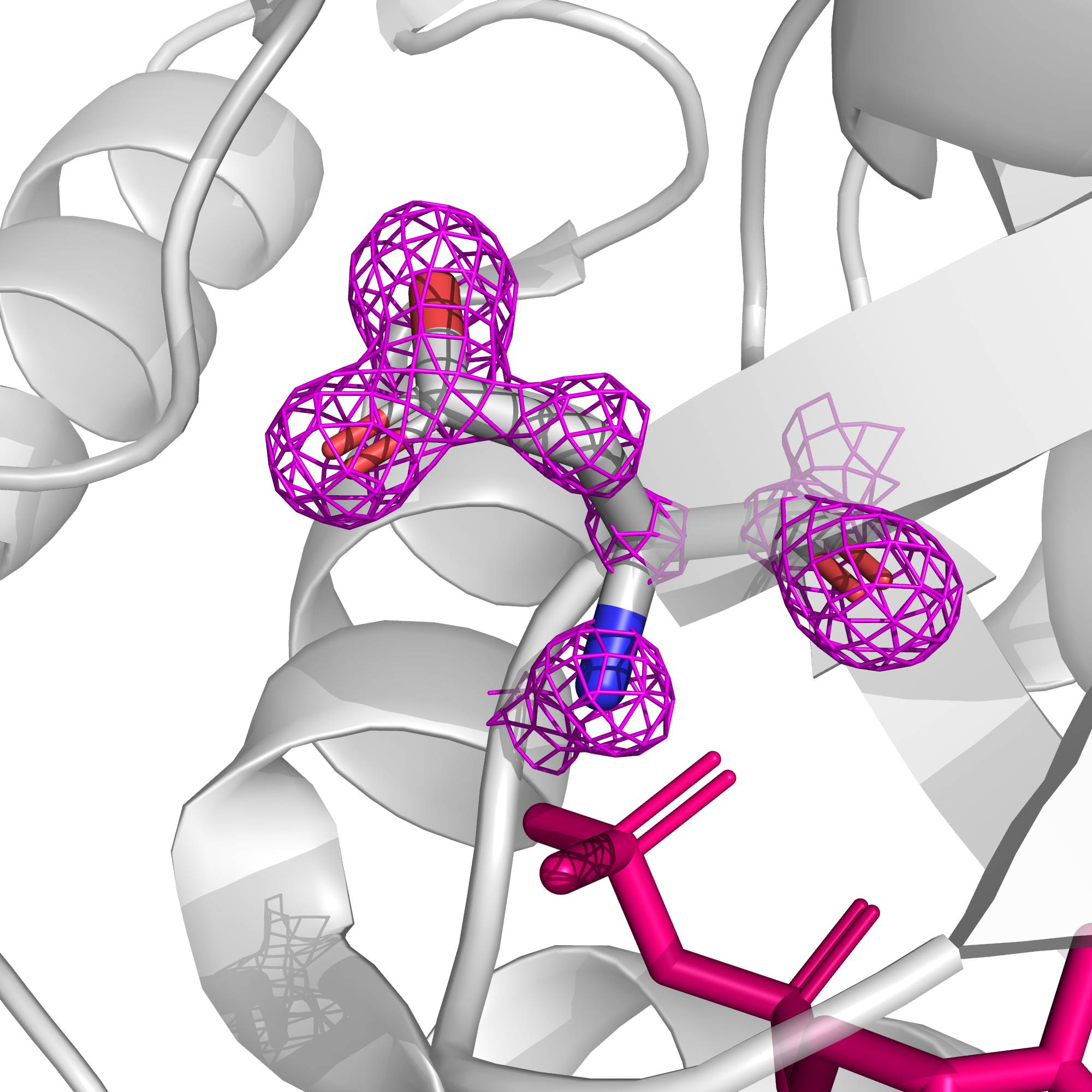
Остаток аспарагиновой кислоты-229 цепи А располагается в домене B недалеко от сайта связывания АДФ. По данным MolProbity, он обладает нестандартными значениями длин и углов связей, а также является довольно редким ротамером. Тем не менее, данные положения атомов достаточно хорошо поддерживаются имеющейся электронной плотностью (рис. 17). Возможно, данный остаток действительно находится в такой необычной конформации (возможно, это может быть связано с его положением на краю бета-поворота, однако данных мало, чтобы что-либо утверждать).
Остаток треонина-88 цепи А достаточно плохо поддерживается электронной плотностью (рис. 18), поэтому, по-видимому, маргинальность данного остатка - следствие ошибки расшифровки.
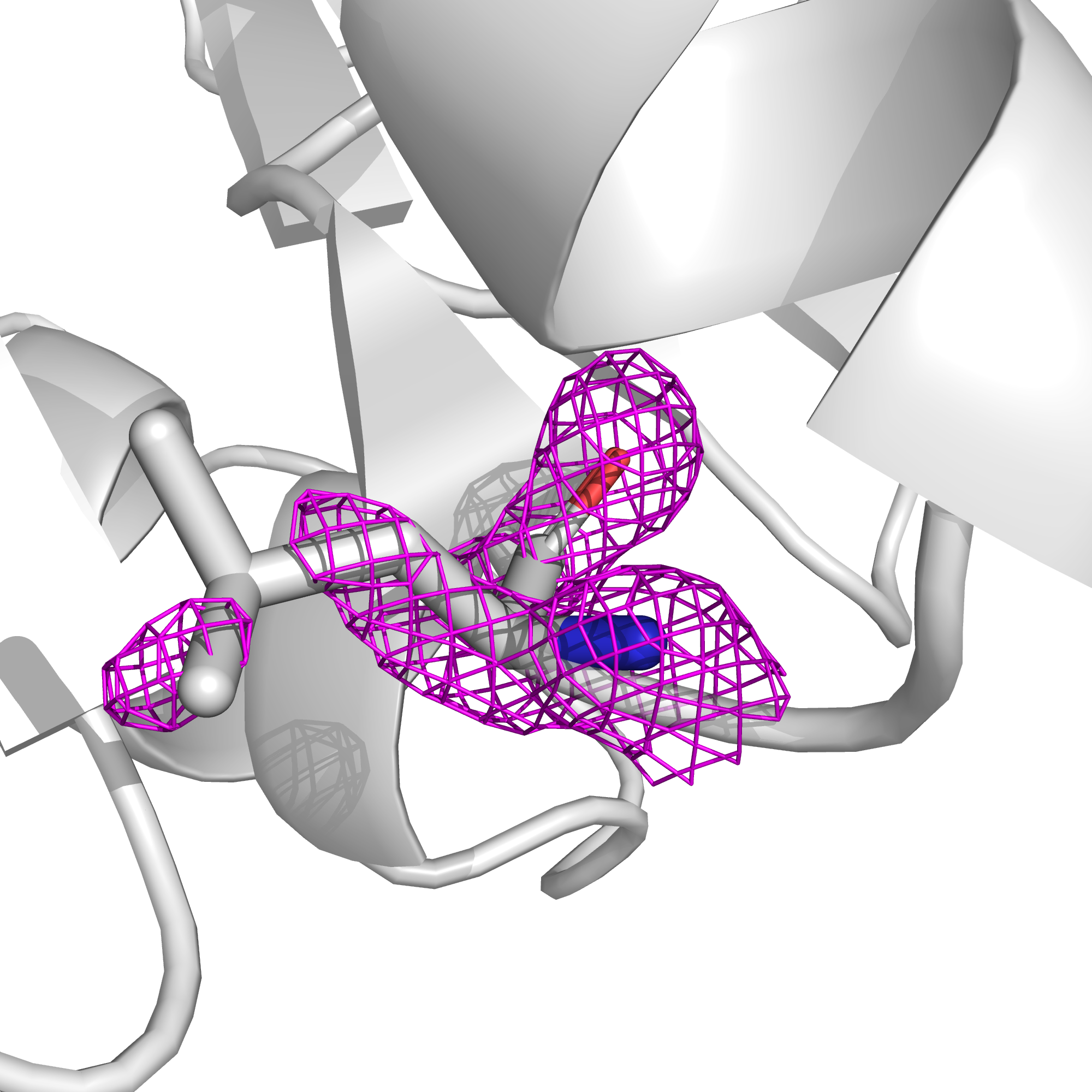
С остатком лейцина-99 цепи А ситуация такая же, как в предыдущем случае: электронная плотность низкая, поэтому маргинальнось может объясняется ошибками расшифровки (рис. 19).
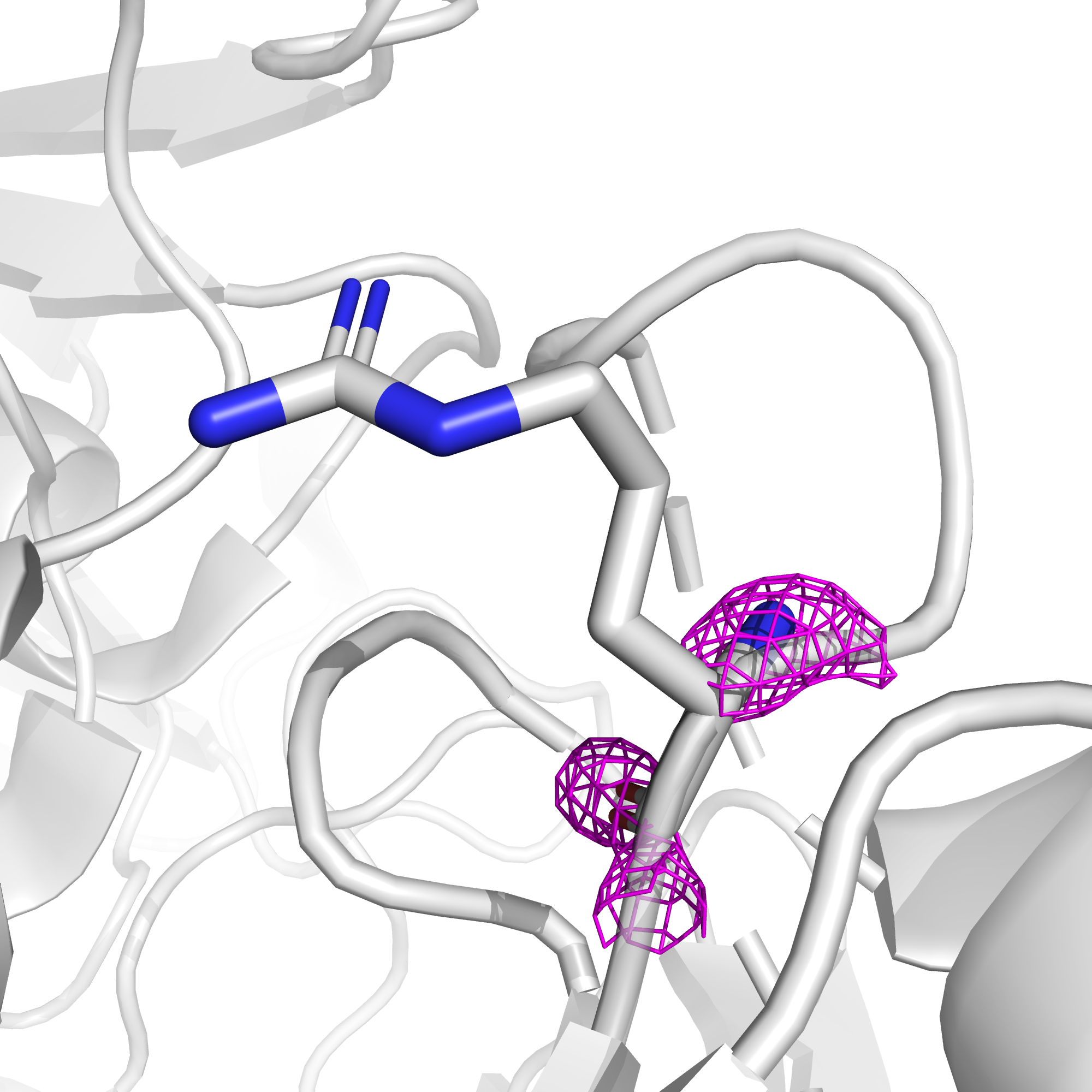
Конформация остататка фенилаланина-307 цепи А достаточно хорошо поддерживается электронной плотностью (рис. 20), и поскольку в принципе MolProbity пишет, что такой ротамер не аутлаер, хотя и принимает пограничные значения, данная конформация, по-видимому, вполне может быть близка к реальной.

Остаток аргинина-191 цепи B представляет собой пример фрагмента структуры совсем плохого качества и совсем не поддерживается электронной плотностью, вследствие чего маргинальность можно считать проблемой расшифровки.
Задание 3. Еще раз про качество в целом
В целом, мы уже в значительной мере изучили качество как структуры в целом и качество функционально значимых участков (задание 1), так и качество отдельных маргинальных остатков (задание 2).
Если мы рассмотрим карман связывания ADP, то увидим там довольно высокое качество остатков: никакие из них не являются маргинальными по данным MolProbity, а электронная плотность для всех из них выглядит довольно убедительно (рис. 22).
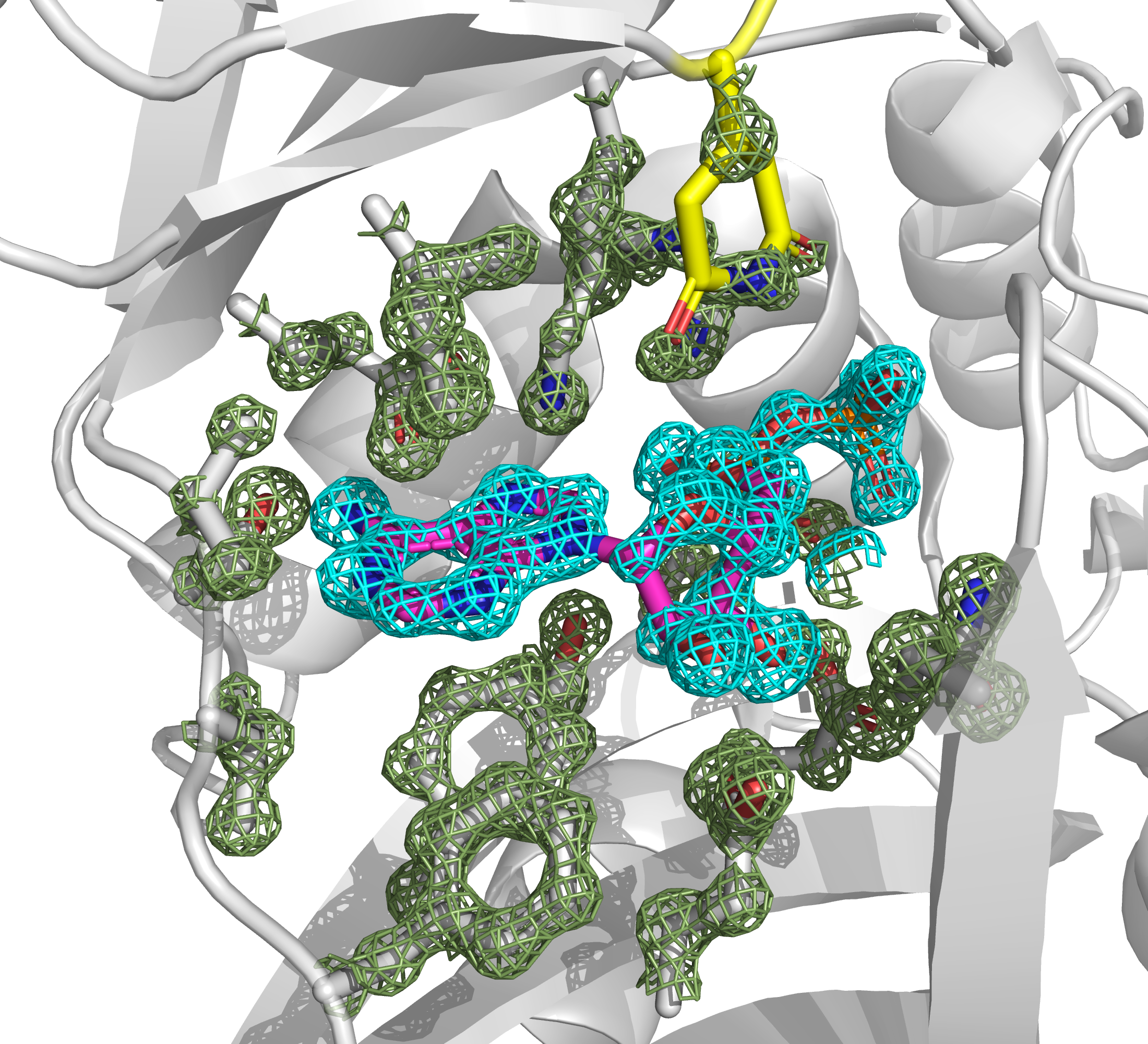
В общем и целом, данная структура, несмотря на относительно большое число маргинальных, а также плохо покрытых электронной плотностью остатков, по-видимому, позволяет изучать связывание ADP, если рассматривать субъединицу I (она же цепь A). Собственно, аналогичный вывод можно было сделать после выполнения первого задания (см. выше).
Задание 4. PDB-REDO
Сервис PDB-REDO предоставляет возможность улучшить качество структуры. Для нашей структуры 1KJQ по некоторым показателям PDB-REDO действительно смог улучшить качество (см. таблицу 1).
| Метрика | Значение (заявленное авторами) | Значение (PDB-REDO) |
|---|---|---|
| Разрешение | 1.05 Å | 1.05 Å |
| R-фактор | 0.188 | 0.156 |
| Rfree | 0.214 | 0.177 |
| Нормальность ротамеров (процентили) | 44 | 78 |
По сhange in density map fit (RSCC) из PDB-REDO, значимо ухудшилось только качество гистидина 51 цепи B, одной молекулы воды и гистидина 54 цепи А. В то же время, многие остатки по RSCC стали лучше.
Если мы посмотрим на приведенные нами выше маргинальные остатки, то увидим, что возможность их улучшения, ожидаемо, зависит от того, насколько хорошо они изначально были вписаны в электронную плотность.
Так, остаток аспарагиновой кислоты-229 цепи А практически не изменился (рис. 23), а остаток треонина-88, вокруг которого электронная плотность была очень низкая, PDB-REDO заметно подправил (рис. 24).
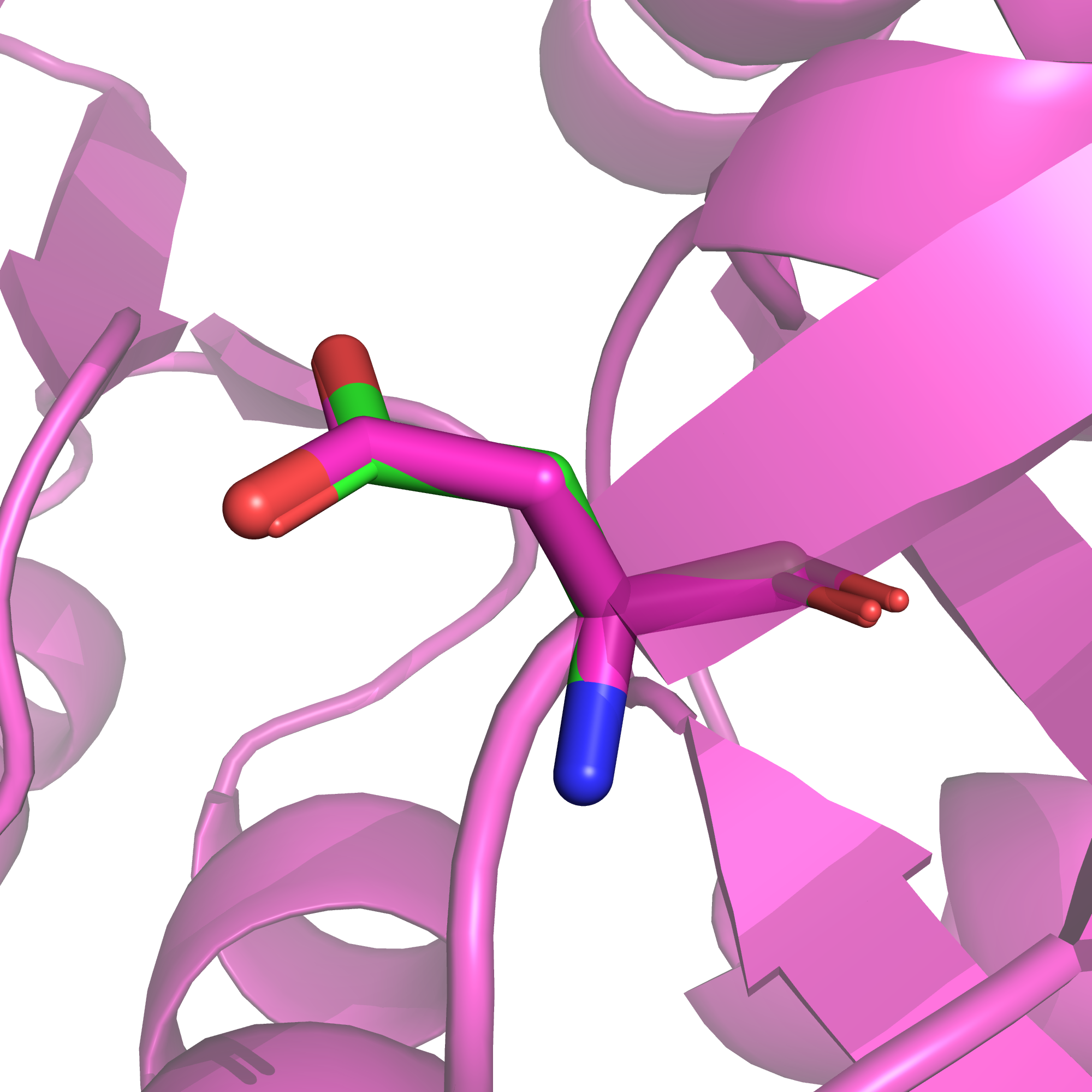
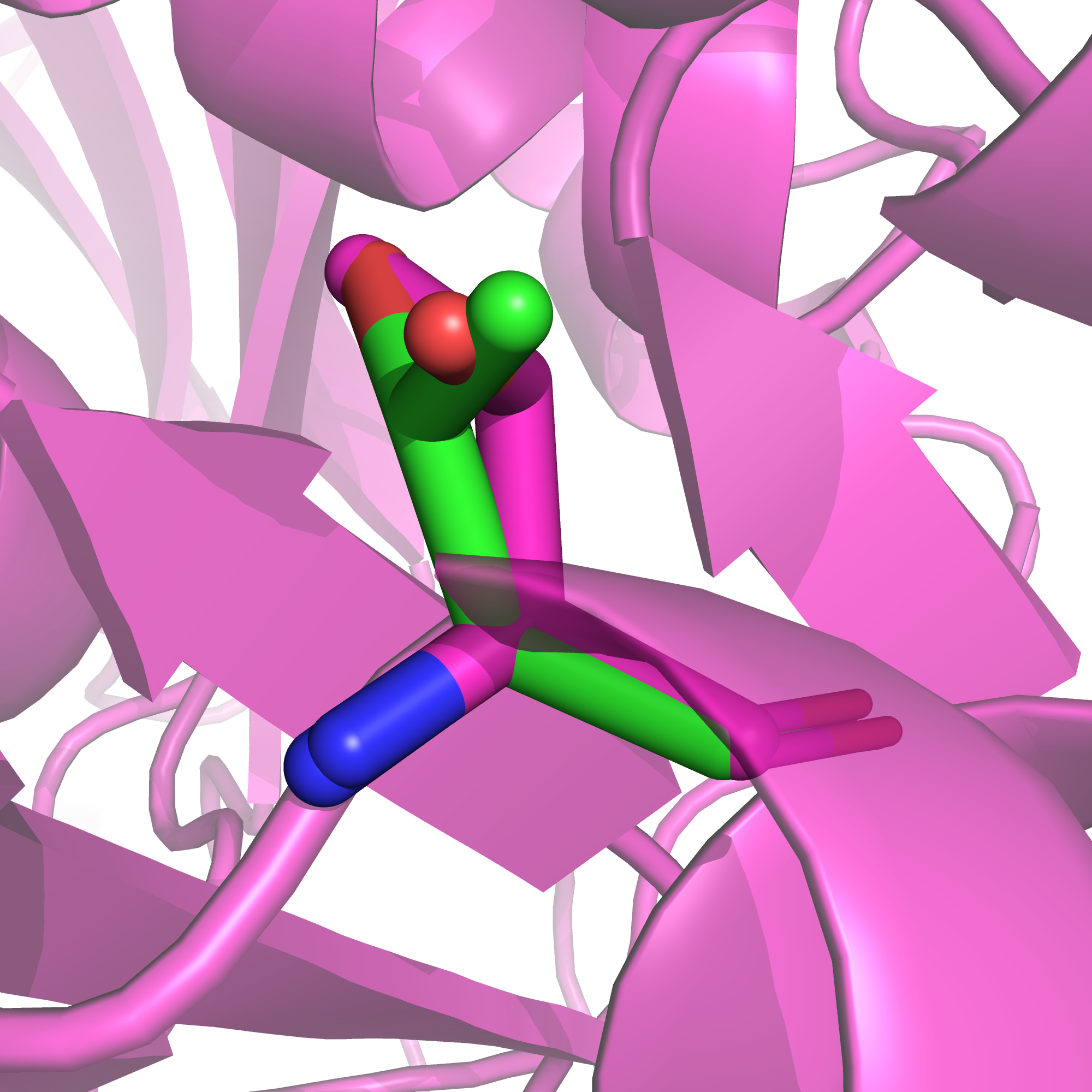
Можно заключить, что структуру из PDB-REDO для изучения белка действительно можно использовать, но не нужно ждать значительных изменений в функционально значимых участках субъединицы I, которые и так, судя по всему, были хорошо вписаны в электронную плотность.
Список литературы
1. Thoden JB, Firestine SM, Benkovic SJ, Holden HM. PurT-encoded glycinamide ribonucleotide transformylase. Accommodation of adenosine nucleotide analogs within the active site. J Biol Chem. 2002 Jun 28;277(26):23898-908. doi: 10.1074/jbc.M202251200. Epub 2002 Apr 12. PMID: 11953435.
2. Vidhi Pareek, Anthony M. Pedley & Stephen J. Benkovic (2021) Human de novo purine biosynthesis, Critical Reviews in Biochemistry and Molecular Biology, 56:1, 1-16, DOI: 10.1080/10409238.2020.1832438