Anna Zheltova
A-, B-, Z- form DNA (A-, B-, Z-формы ДНК)
Complexes of DNA-protein (Комплексы ДНК-белок)
Reading Sanger sequencing (Прочтение последовательностей по Сэнгеру)
Nucleotide databanks (Нуклеотидные банки данных)
Aligning genomes (Выравнивание геномов)
The genes of prokaryotes (Гены прокариот)
The genes of eukaryotes (Гены эукариот)
Search for snp (Поиск полиморфизмов)
Задача: Найти и описать полиморфизмы у пациента
Дано:
1. Чтения экзома, картирующиеся на участок хромосомы человека.Файл с одноконцевыми чтениями в формате fastq (chr9_1.fastq)
2. Хромосомы человеческого генома (сборка версии hg19). Хромосома 9.
Часть I
Этап 0
• Была создана рабочая директория и скопированы файлы, доступные по ссылкам выше.
Этап 1
Анализ качества чтений.
Был сделан контроль качества чтений с помощью программы FastQC.
Программа FastQC, установленная на kodomo, была вызвана командой "fastqc chr9_1.fastq". Версия с графическим интерфейсом была установлена на компьютер.
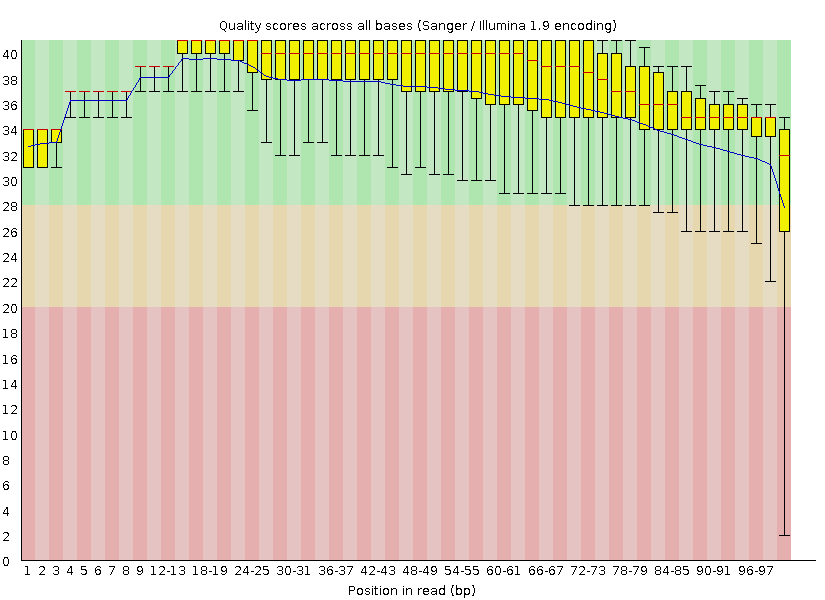
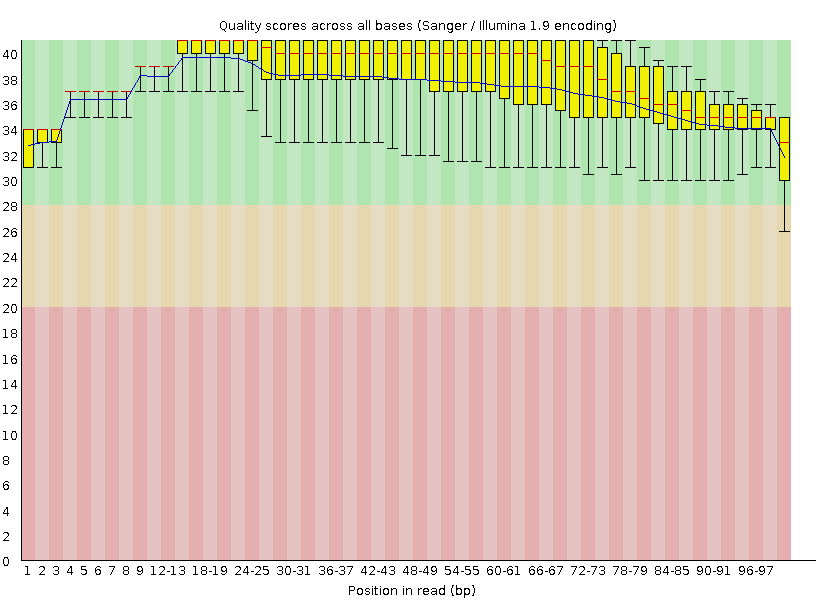
На рисунке показано качество определения основания в каждой позиции рида. На графике видно три полосы, одна из которых – зеленая, другая- желтая, а третья – красная. Если рид попадает в красную область, то его качество низкое, если в зеленую – высокое, а в желтую – среднее.
Этап 2. Очистка чтений
• Сделайте очистку чтений с помощью программы Trimmomatic. Отрежьте с конца каждого чтения нуклеотиды с качеством ниже 20, оставьте только чтения длиной не меньше 50 нуклеотидов.
Очистка была выполнена с через сервер kodomo с помощью программы Trimmomatic с использованием опций TRAILING:20 (отрезает с конца каждого чтения нуклеотиды с качеством ниже 20) и MINLEN:50 (удаляет риды короче 50 нуклеотидов).
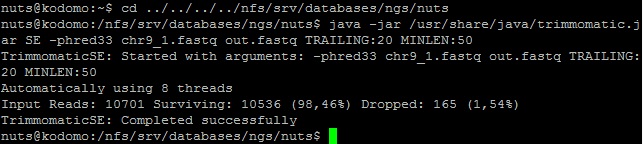
В результате очистки было удалено 165 ридов. В итоге, число оставшихся ридов составило 10536, что соответствует 98,46% от всех исходных ридов.
После чего был сделан контроль качества чтений после очистки с помощью программы FastQC.
Как и ожидалось, были удалены прочтения с низким качеством. Все риды расположены в зеленой области.
Часть II: картирование чтений
Этап 3. Картирование чтений.
• Были откартированы очищенные чтения с помощью программы BWA. Этапы:
o Сначала была проиндексирована референсная последовательность
o Затем было построено выравнивание прочтений и референса в формате .sam.
Этап 4. Анализ выравнивания
• Выравнивание чтений с референсом было переведено в бинарный формат .bam. Для этого использовался пакет samtools.

• Выравнивание чтений с референсом (получившийся после картирования .bam файл) был отсортирован по координате в референсе начала чтения

• Отсортированный .bam файл был проиндексирован.

• Было выяснено, сколько чтений откартировалось на геном
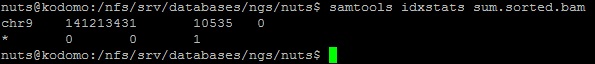
Можно сделать вывод, что откартировалось 10535 ридов. Однако осталось непонятным, куда же делся еще один рид...
Часть III: Анализ SNP
Этап 5. Поиск SNP и инделей.
• Был создан файл с полиморфизмами в формате .bcf;
Однонуклеотидный полиморфизм (ОНП, англ. Single nucleotide polymorphism, SNP) — отличия последовательности ДНК размером в один нуклеотид (A, T, G или C) в геноме (или в другой сравниваемой последовательности) представителей одного вида или между гомологичными участками гомологичных хромосом.
• Был создан файл со списком отличий между референсом и чтениями в формате .vcf.

В итоге было найдено 102 полиморфизма и 6 инделей (различные многонуклеотидные полиморфизмы).
• Были описаны три полиморфизма из .vcf файла.
| Координата | Полиморфизм | Референс (REF) | Чтения (ALT) | Глубина чтений (DP) | Качество чтений(QUAL) |
| 4985879 | Замена | А | G | 30 | 225.009 |
| 5108873 | Замена | C | G | 1 | 6.20226 |
| 5122854 | Индель | AGG | AG | 12 | 96.4609 |
Этап 6. Аннотация SNP.
• С помощью программы annovar были проаннотированы только полученные snp. Были использованы базы данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar.
• Были удалены все индели, после чего файл otl.vcf был переведен в формат .avinput для успешной работы с annovar.
• Аннотация по базе данных RefGene
Было получено два интересующих нас файла: файл RefGeneAnnotation.variant_function (в нем отображены все SNP) и файл RefGeneAnnotation.exonic_variant_function (не трудно догадаться, что в нем содержатся только SNP из экзонов).
Далее предстояло ответить на вопрос: на какие категории делит snp база данных refseq в annovar? Сколько snp попало в каждую группу?
Все возможные категории: exonic, splicing, ncRNA, UTR5, UTR3, intronic, upstream, downstream, intergenic
К exonic относятся 15 snp, к UTR3 - 7, intronic - 78, к downstream – 3, а вот представителей других категорий найдено не было. (Вы сможете ознакомиться со Всеми полученными данными в итоговой таблице)
• Аннотация по базе данных snp138
В этой базе данных можно найти rs для snp. Так как было аннотировано 95 snp, то из этого следует логичный вывод, что 95 snp имеют свои rs. (Вы сможете ознакомиться со Всеми полученными данными в итоговой таблице)
• Аннотирование по базе данных 1000 genomes
В данной базе данных можно найти информацию о частоте встречаемости аллелей. Так как было аннотировано только 84 snp, то только для аннотированных snp (для 84) нам известна частота встречаемости аллелей. (Вы сможете ознакомиться со Всеми полученными данными в итоговой таблице)
• Аннотирование по базе данных Gwas
Было выявлено 8 snp, имеющих клиническое значение. Данное значение было описано в полученном в результате работы программы файле (Вы сможете ознакомиться со Всеми полученными данными в итоговой таблице)
• Аннотирование по базе данных Clinvar
Было найдено 3 snp, имеющих влияние на фенотип. (Вы сможете ознакомиться со Всеми полученными данными в итоговой таблице)
o Хорошее ли покрытие и качество у найденных полиморфизмов?
37 snp имеют покрытие больше 10, что считается хорошим покрытием и имеют высокое качество чтений.
10 snp имеют покрытие от 5 до 10, что считается неплохим покрытием и имеют хорошее качество чтений.
53 snp имеют покрытие меньше 5, что считается плохим покрытием и имеют низкое качество чтений. Из них 37 snp имеют покрытие равное 1, что считается очень плохим покрытием и имеют качество чтений меньше 7, что заставляет задуматься: действительно ли это полиморфизмы.
• В какие гены попали snp?
по базе данных Clinvar snp попали в 2 гена:
o 2 snp попали в ABO
o А для еще одного гена (JAK2) определить, что вызывает snp не удалось.
по базе данных Gwas snp попали в 3 гена:
o 1 snp в JAK2
o 1 snp в IL33
o 6 snp в ABO
• К каким нуклеотидным и аминокислотным заменам привели snp?
Данные приведены в файле exonic_variant_function, полученном при аннотации по refseq. Посмотреть их также можно данном файле.
• Что можно сказать о частоте найденных snp?
Среди аннотированных полиморфизмов:
o 6 с частотами от 1 до 0.8 (очень распространенные).
o 41 с частотами от 0.7 до 0.3 (достаточно распространенные).
o 22 c частотами от 0.3 до 0.1 (не очень распространенные)
o 15 c частотами менее 0.1 (редкие)
• Что можно сказать о клинической аннотации snp?
SNP в гене TNFSF15 вызываeт риск болезни Крона, а SNP в гене вызываeт риск эндометриоза. SNP в гене TNFSF15 определяет риск к заболеванию малярией. Присутствуют SNP, связанные со временем свертывания крови, концентрациями факторов свертываемости и концентрацией гемоглобина в крови, риском венозной тромбоэмболии.
Все команды, использованные в работе, и их описания можно найти в таблице, приведенной выше, Лист 2