Anna Zheltova
A-, B-, Z- form DNA (A-, B-, Z-формы ДНК)
Complexes of DNA-protein (Комплексы ДНК-белок)
Reading Sanger sequencing (Прочтение последовательностей по Сэнгеру)
Nucleotide databanks (Нуклеотидные банки данных)
Aligning genomes (Выравнивание геномов)
The genes of prokaryotes (Гены прокариот)
The genes of eukaryotes (Гены эукариот)
Search for snp (Поиск полиморфизмов)
Для выполнения этого задания был выбран популярный модельный организм нематода Caenorhabditis elegans (C. elegans), используемый для изучения генетического контроля развития и физиологических процессов. C. elegans - свободноживущая нематода (круглый червь) длиной около 1 мм. Исследования этого вида в молекулярной биологии и биологии развития начались в 1974 работами Сиднея Бреннера. Широко используется как модельный организм в исследованиях по генетике, нейрофизиологии, биологии развития, вычислительной биологии. В 1986 году был полностью описан его коннектом. Геном полностью секвенирован и опубликован в 1998 году.
В базе данных NCBI Genome для Danio rerio указаны 2 сборки генома: GCA_000002985.3 и GCA_000975215.1
Число проектов по секвенированию организма и число образцов: 2 сборки генома соответствуют двум проектам (на хромосомном уровне) и двум образцам. Число проектов по транскриптому и неполногеномному секвенированию составляет 594.
Сборка: GCA_000975215.1
1) Описание образца:
• BioSample: SAMN03334911
• Sample name: Caenorhabditis elegans Hawaiian Strain CB4856
• Описание образца и проекта:
| C. elegans Hawaiian strain CB4856 genome sequence 1.0 | С. elegans Гавайский штамм CB4856 последовательность генома 1.0 |
| We have undertaken the construction of a C. elegans Hawaiian strain CB4856 reference genome sequence. The Hawaiian strain, CB4856, was isolated in 1972 by Linda Holden from a pineapple field on the Hawaiian island of Maui (under the name HA8). To complete our reference genome we took advantage of several very deep coverage MPS datasets for the Hawaiian genome, a new de novo assembly program (Chu et al. 2013), end sequences from a fosmid library for the Hawaiian genome (Perkins, J. D., 2010 Comparison of fosmid libraires made from two geographic isolates of Caenorhabditis elegans, University of British Columbia), recently released RNA-seq data and low coverage genome sequence data from 49 recombinant inbred lines (RILs) (Li et al. 2006) and 60 introgression lines (ILs) (Doroszuk et al. 2009). Exploiting these resources and using a variety of software tools, we have modified the C. elegans N2 reference genome to generate a draft reference sequence for the Hawaiian genome.
Using a strategy similar to that employed in the analysis of different Arabidopsis accessions (Gan et al. 2011; Schneeberger et al. 2011), we first aligned the random genomic reads (69.5X coverage composed of 34.7M paired end sequences (104 base sequences) from clones with an insert size of 321 bp with a total of 7220M bases) to the N2 reference genome, identified SNVs and indels, modified the N2 reference accordingly and realigned the reads, repeating the process 19 times to create a first version of the Hawaiian genome (20 cycles total). This process allowed extension of sequence into regions of high divergence, closed large deletions and built sequence into insertions. We used the JR-Assembler v1.0.4 (Chu et al., 2013) to create de novo assemblies of the same sequence reads, assessed their quality using the program REAPR (Hunt et al. 2013), breaking contigs as needed and aligned the resultant contigs to Hawaiian genome. To identify deletions previously missed, we scanned the genome for regions devoid of coverage, merging adjacent regions if they were only separated by short segments of either very low coverage or repeated sequences. For regions flanked by adjacent segments of the de novo assembled contigs, we used the contig to close the gap. To confirm that such segments were properly placed in the genome, we used the RIL data to establish their chromosomal location. The result is an initial draft reference Hawaiian genome with a total length of 97Mb. Regions of excess coverage (>99x) suggest that we have failed to represent some duplicated segments, which total some 0.5 Mb in length. Also, the de novo assembly generated 22 contigs of 16kb total length that we were unable to locate in the reference. We included only the 9 that were at least 500 bases in length. Credits: Illumina Production sequencing - Leonid Kruglyak/Erik C. Andersen, Princeton University, Princeton, NJ USA. Fosmid sequencing - Don Moerman, University of British Columbia, Canada. Sequence assembly and data integration for creation of chromosomal files - Owen A. Thompson and Robert H. Waterston, Department of Genome Sciences at University of Washington School of Medicine, Seattle, WA USA |
Ученые решили сконструировать референсный геном последовательности С. elegans Гавайского штамм CB4856. Гавайский штамм, CB4856, был выделен в 1972 году Линдой Холден с ананасного поля на Гавайском острове Мауи (под именем HA8). Чтобы завершить референсный геном ученые воспользовались несколькими очень масштабными MPS базами данных для Гавайского генома, новая de novo сбора программы (Chu et al. 2013), конечные последовательности из «fosmid» библиотеки для Гавайской генома (Perkins, J. D., 2010 Comparison of fosmid libraires made from two geographic isolates of Caenorhabditis elegans, University of British Columbia), недавно выпущенные РНК-seq данные и низкий охват генома последовательности данных из 49 рекомбинантных инбредных линий (RILs) (Li et al. 2006) и 60 интрогрессионных линий (ILs) (Doroszuk et al. 2009). Эксплуатируя эти ресурсы и используя различные программные средства, мы видоизменили С. elegans Н2 референсный геном чтобы создать проект эталонной последовательности для Гавайского генома. Используя стратегию аналогичную той, которую использовали при анализе различных образцов Арабидопсиса (Gan et al. 2011; Schneeberger et al. 2011), ученые сначала выравнивали случайные считывания (прочтения) геномов (69.5х покрытия, состоящие из 34.7м спаренных последовательностей (104 базовых последовательностей) от клонов со вставкой 321 bp с в общей сложности 7220M баз) с N2 эталонным геномом, определены SNVs и indels,
Соответственно изменены N2 эталонный и перестроено чтение, повторяя процесс 19 раз, чтобы создать первую версию Гавайского генома (всего 20 циклов). Этот процесс допускает расширение последовательности в регионах с высоким уровнем дивергенции, закрывающее крупные делеции и вставки в построенной последовательности. Ученые использовали линии JR-Assembler v1.0.4 (Chu et al., 2013) для создания de novo сборок одной и той же прочтенной последовательности, оценивая их качество с использованием программы REAPR (Hunt et al. 2013), разрушая континги по мере необходимости и выравнивая результирующие континги для Гавайского генома. Для выявления ранее пропущенных делеций, ученые сканировали геном для регионов, лишенных покрытия, слияния соседних регионов, если они разделяются лишь короткими сегментами, либо очень низкий охват или повторяющиеся последовательности. Для регионов между смежными сегментами de novo были собраны континги, ученые использовали континги, чтобы закрыть разрыв. Чтобы подтвердить, что такие сегменты были правильно размещены в геноме, мы использовали RIL данные для установления их хромосомного расположения. В результате получился первоначальный проект эталонного Гавайского генома общей протяженностью 97Mb. Регионы избыточного покрытия (>99x) предлогают, что мы не смогли представлять некоторые повторяющиеся сегменты, которые составляют около 0.5 Мб в длину. Кроме того, de novo сборка, генерируется 22 контингами общей длиной 16kb, что мы были не в состоянии найти в справочнике. Мы включили только те 9, которые были по меньшей мере 500 оснований в длину. Illumina Production sequencing - Leonid Kruglyak/Erik C. Andersen, Princeton University, Princeton, NJ USA. Fosmid sequencing - Don Moerman, University of British Columbia, Canada. Sequence assembly and data integration for creation of chromosomal files - Owen A. Thompson and Robert H. Waterston, Department of Genome Sciences at University of Washington School of Medicine, Seattle, WA USA |
• Проект: PRJNA275000
1) Число контингов: 16
2) Число скэффолдов: 15
3) Таблица контигов/скэффолдов
4) N50: 14890789 п.о.
5) L50: 3
6) Самый длинный контиг: 20182852 п.о.
7) Самый короткий контиг: 607 п.о.
8) Ссылка на последовательность одного из контигов
2. Составьте таблицу митохондриальных генов мха Bartramia pomiformis

1) Запрос: ((Bartramia pomiformis [orgn]) and mitochondrion) and complete genome Было получено два результата. Первый результат из банка RefSeq, а второй из GenBank. 
Для анализа была выбрана первая запись, так как она из базы данных проверенных геномов. Информация о митохондриальном геноме: 
Всего в этой записи 67 генов, 40 кодируют белки, 24 - тРНК, 3 - рибосомальные РНK.
3. Опишите десять ключей, используемых в таблицах особенностей
1) LTR - длинные концевые повторы, последовательности непосредственно повторяют оба конца определенной последовательности, что, как правило, можно найти в ретровирусах.
E.coli/Tn4521 left junction

2) regulatory – участок последовательности, участвующий в регуляции транскрипции или трансляции.
Human cytokine (SCYA2) gene, exon 1

3) exon - область генома, которая кодирует часть сплайсированной мРНК, рРНК и тРНК; может содержать 5'UTR, все CDSs и 3' - UTR;
Human cytokine (SCYA2) gene, exon 1

4) intron - сегмент ДНК, который транскрибируется, но удаляется из транскрипта путем соединять воедино последовательностей (экзонов) по обе стороны от него.
Human cytokine (SCYA2) gene, exon 1

5) mobile_element - участок генома, содержащий подвижные элементы.
Hyperthermophilic Archaeal Virus 2, complete genome

6) stem_loop - шпилька; двойной спиральный регион, сформированный сопряжением оснований между смежными (инвертированными) комплементарными последовательностями единой цепи РНК или ДНК.
Mytilus galloprovincialis histone H4 (H4), histone H2B (H2B), histone H2A (H2A), histone H3 (H3), and histone H1 (H1) genes, complete cds; and 5S ribosomal RNA and 5S ribosomal RNA genes, complete sequence

7) transit_peptide – пептид, кодирующий последовательность N-концевого домена закодированных в ядре органеллы белка; этот домен участвует в посттрансляционном импорте белка в органеллу.
Rattus norvegicus mRNA for mitochondrial aconitase (nuclear aco2 gene)

8) polyA_site - сайт РНК, к которому присоединяются остатки аденина вследствие посттранскрипционного полиаденилирования.
Cicer arietinum mRNA for hypothetical protein (294 gene), polyA site
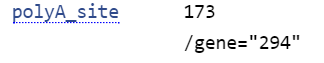
9) misc_feature - необычный участок, который не описывается никаким известным ключом; новая или редкая характеристика.
Mus musculus centromere protein A (Cenpa), transcript variant 2, mRNA

10) misc_recomb - сайт любого обобщенного, конкретного узла или события репликативной рекомбинации, где есть обрыв и воссоединение дуплексных ДНК, которые не могут быть описаны другими рекомбинантными ключи или квалификаторами исходного ключа.
Cloning vector pENTR/Zeo-H2A, complete sequence
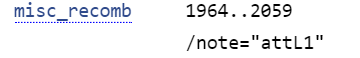
4. (blastn) Установите какому гену принадлежит последовательность, полученная в практикуме 6, и таксономию организма
Был использован BLASTN.
Пять лучших находок:
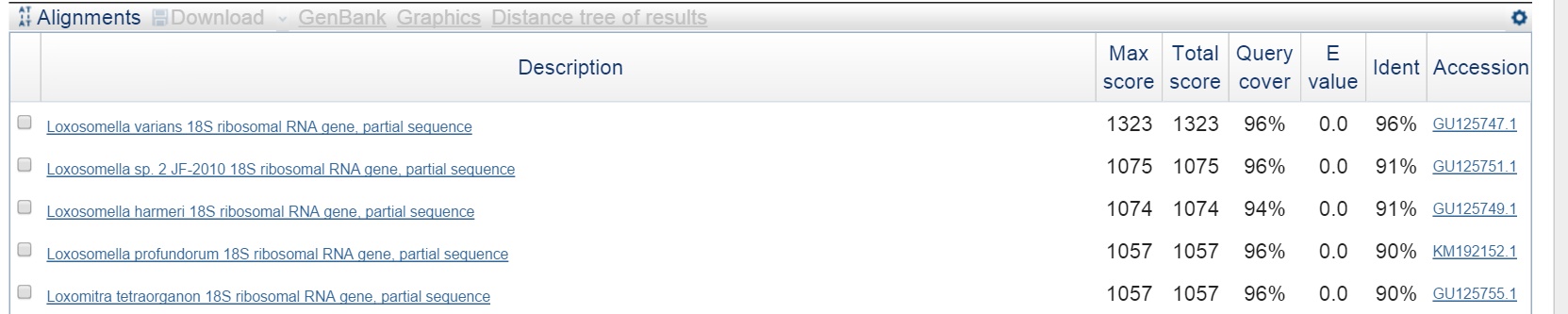
Как видно из рисунка, E-value находок составляет ноль. Это означает, что данные находки не случайны.
На основании пяти представленных находок можно сделать следующие выводы:
• Все последовательности принадлежат 18S ribosomal RNA gene. Это означает, что полученная в практикуме №6 последовательность кодирует 18S ribosomal RNA gene.
18S рибосомальная РНК.
18S рРНК (сокращенно рРНК 18S) является частью рибосомальной РНК. 18S рРНК является составной частью малой субъединицы эукариотической рибосомы (40S). рРНК 18S является структурной РНК для небольшого компонента эукариотических цитоплазматических рибосом, и, таким образом, одним из основных компонентов всех эукариотических клеткок. Это эукариотический ядерный гомолог 16S рибосомальной РНК прокариот и митохондрий.
Гены, кодирующие рРНК 18S называют 18S-рДНК. Последовательность данных этих генов широко используется в молекулярном анализе для реконструкции эволюционной истории организмов, особенно позвоночных, так как её медленная эволюционная скорость делает её пригодной для восстановления древних расхождений.
Использание в филогенезе.
Ген малой субъединицы (SSU) 18S рРНК является одним из наиболее часто используемых генов в филогенетических исследованиях и важным маркером для случайной целевой полимеразной цепной реакции (ПЦР) в скрининге окружающего биоразнообразия. В целом, генные последовательности рРНК легко доступны в результате высоко консервативных фланкирующих регионов, позволяющих использовать универсальные праймеры. Их расположение, повторяющееся в геноме, обеспечивает избыточное количество матричной ДНК для ПЦР, даже в самых маленьких организмах. Ген 18S является частью рибосомной функциональной зоны и подвергается подобным селективным силам во всех живых существах. Таким образом, когда были опубликованы первые крупномасштабные филогенетические исследования, основанные на 18S последовательностей - в первую очередь, филогения животного царства и др. Ген отмечается как главный кандидат для использования в реконструкции древа жизни многоклеточных.
• Таксономия лучших находок: 4 из 5 находок принадлежат одному роду, в то же время все пять находок относятся к одному семейству.
Classification: Biota > Animalia (Kingdom) > Entoprocta (Phylum) > Solitaria (Order) > Loxosomatidae (Family)
o > Loxosomella (Genus) – 4 находки
• Species Loxosomella varians Nielsen, 1964 – первая находка
• Species Loxosomella sp. 2 JF-2010 – вторая находка
• Species Loxosomella harmeri (Schultz, 1895) – третья находка
• Species Loxosomella profundorum Borisanova, Chernyshev, Neretina & Stupnikova – четвертая находка
o > Loxomitra (Genus) – 1 находка
• Species Loxomitra tetraorganon Iseto, 2002 – пятая находка
Мой организм относится к семейству Loxosomatidae. Так как четыре лучших находки из 5 принадлежат организмам из рода Loxosomella, то можно предположить, что данный организм относится к этому роду.
Было построено выравнивание входной последовательности с находками: файл