Anna Zheltova
A-, B-, Z- form DNA (A-, B-, Z-формы ДНК)
Complexes of DNA-protein (Комплексы ДНК-белок)
Reading Sanger sequencing (Прочтение последовательностей по Сэнгеру)
Nucleotide databanks (Нуклеотидные банки данных)
Aligning genomes (Выравнивание геномов)
The genes of prokaryotes (Гены прокариот)
The genes of eukaryotes (Гены эукариот)
Search for snp (Поиск полиморфизмов)
Упражнения:
1) (seqret) Несколько файлов в формате fasta собрать в единый файл
Сначала с помощью команды ls > list был создан файл с названиями записей.
Команда seqret @list.txt sequences.fasta
На выходе был получен файл с обоими последовательностями, названия которых были в файле list.
2) (seqretsplit) Один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы
seqretsplit sequences.fasta
В файле sequences.fasta (полученном в предыдущем упражнении) были записаны последовательности. Результатом выполнения программы было разделение этих последовательности на отдельные файлы, т.е. каждой последовательности соответствовал один файл.
3) (seqret) Из файла с хромосомой в формате .gb вырезать три кодирующих последовательности по указанным координатам "от", "до" и сохранить в одном fasta файле
Каждая последовательность хромосомы имеет свой АС.
seqret @list1.txt out.fasta
Содержание файла @list1.txt:
genbank:АС[от:до:ориентация]
genbank:АС[от:до]
genbank:АС[от:до]
Например:
genbank:AE017042[10781:11470:r]
genbank:AE017042[9277:10701]
genbank:AE017042[8131:9057]
В результате получили файл (out.fasta), в котором были записаны три кодирующие последовательности с заданными характеристиками.
4) (transeq) Транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода. Результат - в одном fasta файле.
Был использован файл out.fasta из предыдущего задания.
transeq out.fasta
В результате получили файл с 3 аминокислотными последовательностями, соответствующими исходным.
5) (transeq) Транслировать данную нуклеотидную последовательность в шести рамках.
transeq cod.fasta out_frames.fasta –frame 6
В результате получили файл frames.fasta оказалось 6 белковых последовательностей, полученных при транслировании последовательности cod.fasta в 6 рамках.
Сравните аннотации генов белков в одной хромосоме бактерии или археи с трансляциями длинных открытых рамок считывания
1. Получите список трансляций открытых рамок с помощью команды getorf пакета EMBOSS
Была выбрана архея Methanopyrus kandleri AV19 последовательность в fasta-формате. GenBank: AE009439.1
Получили файл ae009439.orf
2. Получите список координат и ориентаций найденных открытых рамок с помощью infoseq

Полученная, отредактированная таблица
3. Получите список аннотированных генов белков
• таблица аннотированных генов белков в формате Excel.
• файл с последовательностями белков в формате fasta
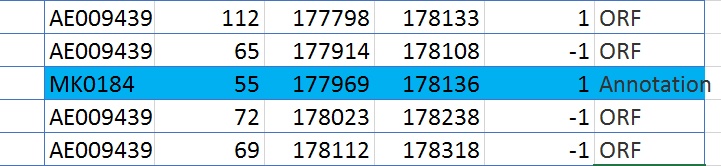
Было проведено сравнение таблицы аннотированных белков и таблицы открытых рамок (файл «Объединенная таблица»). В ходе сравнения было выявлено:
• Открытых рамок было выявлено намного больше (14810), чем белков аннотировано (1687)
• точное соответствие начала и конца кодирующих последовательностей мне не встретилось
• В большинстве случаев был обнаружен сдвиг рамки считывания на три нуклеотида в начале и/или в конце.
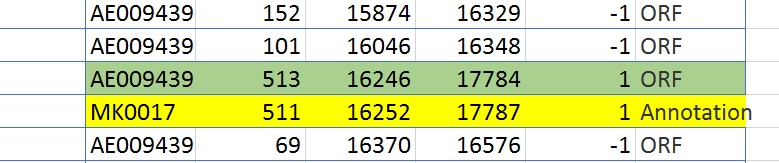
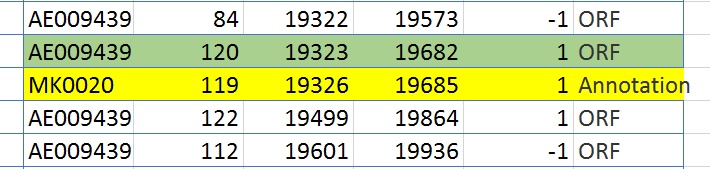
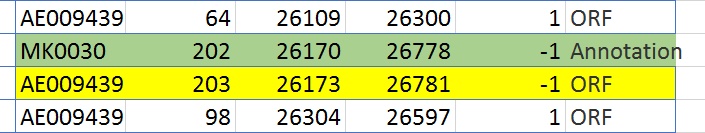
• Были выявлены случаи, когда начало практически совпадало, но не наблюдалось совпадение конца
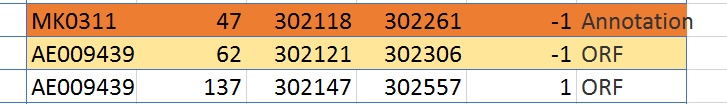
• С помощью программы getorf не были найдены некоторые белки. Вероятно, это связано с тем, что установленный порог длины соответствовал белкам размером более 60 а.о.