Anna Zheltova
Fourth term (Четвертый семестр):
Molecular phylogeny (Молекулярная филогения)
Phylogeny reconstruction (реконструкция филогении)
Enzymes and metabolic pathways. KEGG database. (Ферменты и метаболические пути. База данных KEGG)
Membrane proteins(Мембранные белки)
Задание 1. Сравнить состав систем рестрикции-модификации, закодированных в двух штаммах одного вида
Задача: сравнить предполагаемые (по избеганию сайтов) наборы систем рестрикции-модификации (Р-М) в полном геноме бактерии из NCBI и наборе контигов того же вида из метагенома кишечника человека.
Материал:
Геном из БД NCBI
Фрагменты генома из метагенома кишечника человека
Этап 1. Найдите избегаемые сайты рестрикции в геноме выданной бактерии или археи
Мне досталась бактерия Bifidobacterium breve (идентификатор GenBank CP000303.1)
С помощью веб-сервиса , используя метод Карлина, был посчитан ожидаемый контраст и количество всех сайтов из файла , который содержит список почти всех известных сайтов систем Р-М типа II (основной тип), в геноме полученной бактерии
Полученный результат содержится в файле .
Были найдены все сайты, для которых контраст меньше чем 0.78 (это порог, чтобы отличие от 1 можно было считать значительным). Таких сайтов всего 1 (Сайт CTAG). Информация о данном сайте представлена в файле .
Этап 2. Найдите избегаемые сайты рестрикции в наборе контигов из метагенома
С помощью веб-сервиса , используя метод Карлина, был посчитан ожидаемый контраст и количество всех сайтов из файла , который содержит список почти всех известных сайтов систем Р-М типа II (основной тип), в наборе контигов из метагенома полученной бактерии
Полученный результат содержится в файле .
Были найдены все сайты, для которых контраст меньше чем 0.78 (это порог, чтобы отличие от 1 можно было считать значительным). И снова таких сайтов только 1 (уже встречавшийся ранее сайт CTAG). Информация о данном сайте представлена в файле.
Сравнив две полученные таблицы, можно сказать, что:
1. Как в геноме, так и в наборе контигов из метагенома присутствует только один сайт с контрастом меньше, чем 0.78
2. Данный сайт для генома и контигов из метагенома совпадает.
3. В данной таблице приведен объединенный результат по двум этапам. Сайты совпадают.
Этап 3. Сравните полученные списки избегаемых сайтов
Избегаемых сайтов найдено только в полном геноме - 1, только в контигах - 1, Данный сайт совпадает.
Обе бактерии содержали одинаковое количество систем Р-М, возможно потому, что данные организмы очень близкие. Один из них жил в кишечнике человека, а второй (полный геном)… На сайте NCBI в соответствующей записи базы данных Nucleotide этой информации мне найти не удалось, но предположу, что данный организм жил в тех же (или мало отличающихся) условиях.
Задание 2. Найти последовательности Шайн-Дальгарно в геноме бактерии, полученной в первом семестре.
Бактерия Methanopyrus kandleri AV19 была найдена в БД Assembly на NCBI . Со страницы последовательности в GeneBank (GenBank: AE009439.1) был скачан fasta файл с хромосомой. Так же был скачан файл с особенностями (features) , среди них есть CDSs.
Файл с Features был преобразован в .xls формат с координатами кодирующих последовательностей. Для этого был использован скрипт. Получившийся файл
Было выбрано 200 "хороших" кодирующих последовательностей. "Хорошая" значит есть надежда, что ген хорошо аннотирован: не гипотетический, достаточно длинный (скажем, более 300 п.н.). Выбранные последовательности представлены в данной таблице (лист «выбранные последовательности»). Из выборки были удалены все гипотетические и не имеющие характеристик последовательности, а также последовательности меньшей длины, чем 300 п.н. Из них было выбрано 200 последовательностей.
Последовательность Шайна — Дальгарно (англ. Shine-Dalgarno sequence, Shine-Dalgarno box) — сайт связывания рибосом на молекуле мРНК прокариот, обычно на расстоянии около 10 нуклеотидов до стартового кодона AUG . Описана австралийскими учёными Джоном Шайном и Линн Дальгарно. Шайн-Дальгарно последовательность у прокариот определяет эффективность связывания рибосом в участке инициации трансляции. Последовательность Шайна-Дальгарно - это пуриновая последовательность AGGAGG в бактериальной мРНК , облегчающая ее связывание с рибосомой
Как было выяснено ранее, искомая последовательность состоит из 6 нуклеотидов и располагается на расстоянии около 10 нуклеотидов от стартового кодона. В связи с этим предполагается правильным взять участок [-16;0] у выбранных генов. Однако, принимая во внимание рекомендацию по поиску и учитывая то, что расстояние, указанное в литературе, примерно, было решено расширить область поиска до [-20;0] (лист «поиск»)
Далее с помощью скрипта из файла с полным геномом были вырезаны участки последовательностей, соответствующие координатам последовательностей, предположительно включающих Ш-Д. Результат .
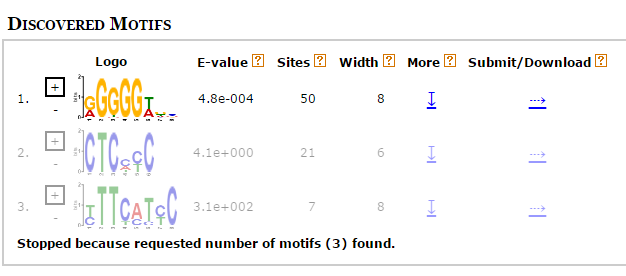
Далее использовалась онлайн версия программы MEME . Для поиска мотивов были использованы: файл с последовательностями, предположительно содержащими Ш.-Д. , длина мотива при поиске составляла от 4 до 10 нуклеотидов, поиск только по данной цепи, ожидаемое количество мотивов в последовательности – от 0 до 3 (поиск для нахождения 3-х разных мотивов, чтобы сравнить e-value).
Параметры, с которыми производила запуск программы:
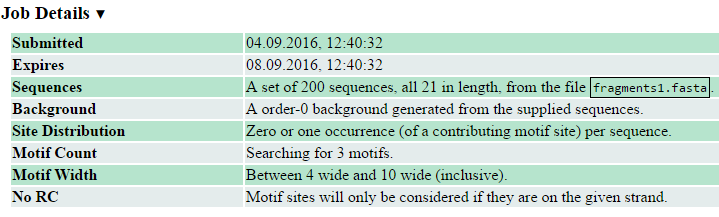
При анализе выдачи МЕМЕ была выявлена проблема: e-value первого найденного мотива намного меньше, чем e-value второго и третьего, однако оно все-равно большое.
Изучив распределение количества находок в зависимости от позиции начала SD, я пришла к выводу, что они все начинались и заканчивались в рамках выбранного диапазона, что означает, что проблема явно заключается не в выбранном диапазоне. Поэтому поиск решено было повторить.
Было выбрано 237 "хороших" кодирующих последовательностей. "Хорошая" значит есть надежда, что ген хорошо аннотирован: не гипотетический, достаточно длинный (скажем, более 300 п.н.). Выбранные последовательности представлены в данной таблице (лист «выбранные последовательности2»). Из выборки были удалены все гипотетические последовательности, а также последовательности меньшей длины, чем 300 п.н. Из них было выбрано 237 самых длинных.
Как было выяснено ранее, искомая последовательность состоит из 6 нуклеотидов и располагается на расстоянии около 10 нуклеотидов от стартового кодона. В связи с этим предполагается правильным взять участок [-16;0] у выбранных генов. Однако, принимая во внимание рекомендацию по поиску и учитывая то, что расстояние, указанное в литературе, примерно, было решено расширить область поиска до [-20;0] (лист «поиск2»)
Далее с помощью скрипта из файла с полным геномом были вырезаны участки последовательностей, соответствующие координатам последовательностей , предположительно включающих Ш-Д Результат .
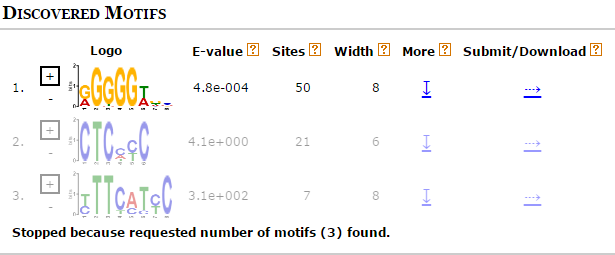
Далее использовалась онлайн версия программы MEME . Для поиска мотивов были использованы: файл с последовательностями, предположительно содержащими Ш.-Д. , длина мотива при поиске составляла от 4 до 10 нуклеотидов, поиск только по данной цепи, ожидаемое количество мотивов в последовательности – от 0 до 3 (поиск для нахождения 3-х разных мотивов, чтобы сравнить e-value).
И снова при анализе выдачи МЕМЕ была выявлена проблема: e-value первого найденного мотива намного меньше, чем e-value второго и третьего, однако оно все-равно большое.
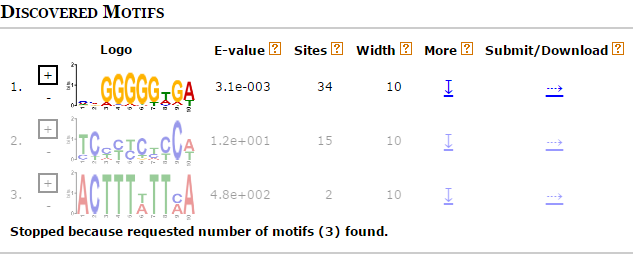
Найденный мотив для данных генов совпадает с найденным ранее мотивом. На основании этого и, принимая во внимание то, что последовательность Шайна-Дальгарно - это пуриновая последовательность, будем считать что первый мотив и есть искомая последовательность (возможно, что для данного вида сама последовательность Шайна-Дальгарно не канонична (т.е. не классическая AGGAGG, что вполне возможно, так как я рассматривала архею) или же имеется некая недопредставленность внутренних последовательностей Шайна-Дальгарно.
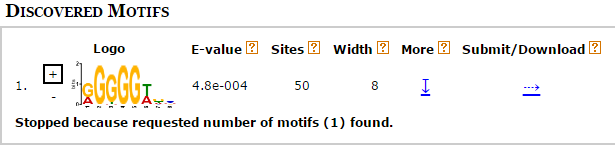
И так, было решено вернуться к первой выдаче программы МЕМЕ и проанализировать ее .
Три разных мотива, найденные MEME.
Лого мотивов, найденных программой MAST
Я провела поиск в MEME с теми же параметрами, но только для лучшего мотива.
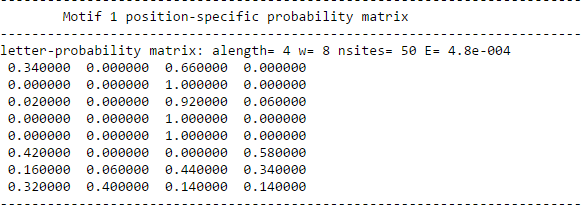
В результате была получена позиционная матрица весов.
![]()
Позиционная матрица весов для первого мотива:
Поиск ШД во всем геноме с помощью PWM, построенной MEME
Поиск по регионам перед всеми генами проводился уже с помощью ресурса FIMO
На основе PWM , построенной ранее был произведен поиск найденного мотива. Поиск мотива был выполнен для всех генов археи (всего их 1691) (участки от -20 до 1 нуклеотида от старта трансляции).
Параметры, с которыми была запущена FIMO

Было найдено 1517 мотивов для p-value меньше 0.01
Выдача FIMO в html
Найденных мотивов меньше, чем исходных последовательностей, что скорее всего можно объяснить тем, что среди исходных последовательностей много случайных, а также есть вероятность того, что не было найдено какое-то количество неслучайных (реально существующих) последовательностей.
Задание 3. Определите сайты связывания данного транскрипционного фактора в данном участке хромосомы человека
Исходный файл: chipseq_chunk10.fastq .
С помощью программы FastQC был сделан контроль качества прочтений
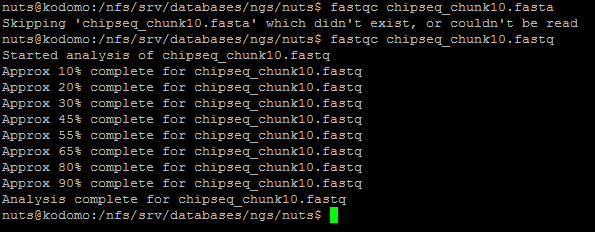
Результат работы программы представлен в файлах chipseq_chunk10_fastqc.html и chipseq_chunk10_fastqc.zip.
На рисунке показано качество определения основания в каждой позиции рида. На графике видно три полосы, одна из которых – зеленая, другая- желтая, а третья – красная. Если рид попадает в красную область, то его качество низкое, если в зеленую – высокое, а в желтую – среднее.
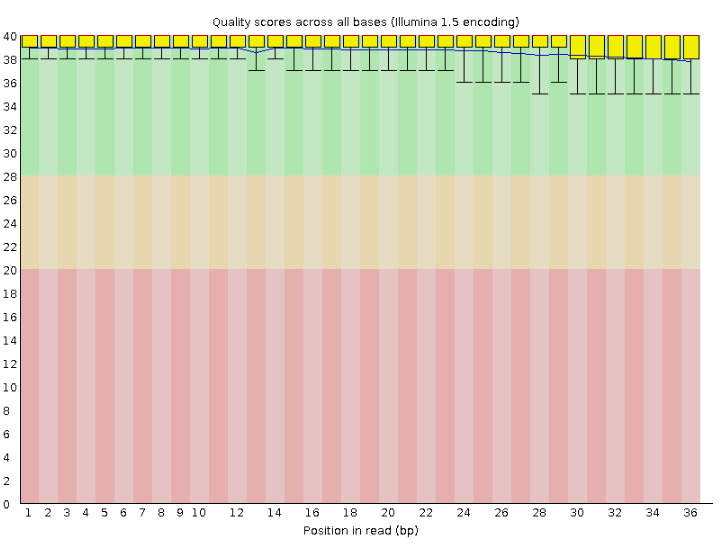
Как следует из представленного изображения, качество чтений высокое, поэтому очистка программой Trimmomatic не требовалась.
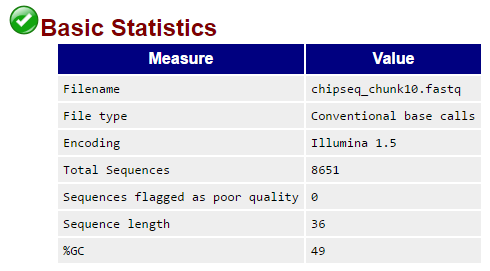
Общее число чтений: 8651, длина ридов: 36.
Далее было проведено выполнено картирование прочтений на геном человека hg19.
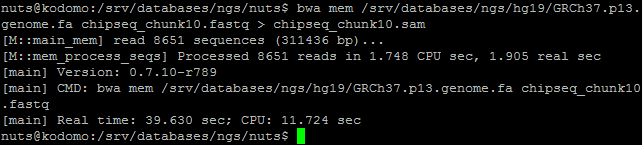
Далее был проведен анализ полученного выравнивания из файла в формате .sam при помощи команд из таблицы:
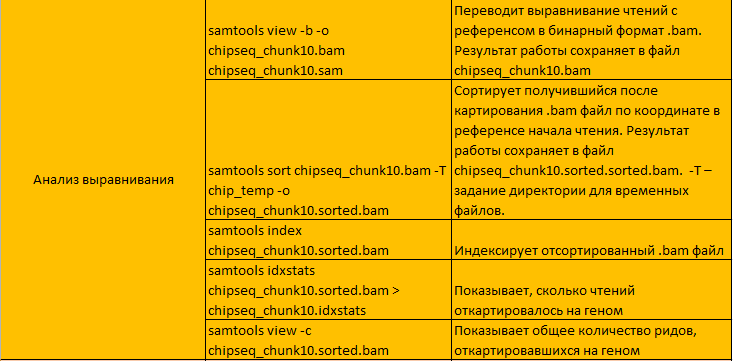

Откартировались все 8651 рид.
После былв запущена программа для поиска пиков.
macs2 callpeak -t chipseq_chunk10.sorted.bam
Пиков оказалось мало, поэтому данная программа запущена с другими параметрами: macs2 callpeak –t chipseq_chunk1.sorted.bam --nomodel -nchunk10
Были получены следующие файлы chunk10_peaks.narrowPeak,chunk10_peaks.xls , chunk10_peaks.xls , chunk10_summits.bed , chunk10_summits.bed .
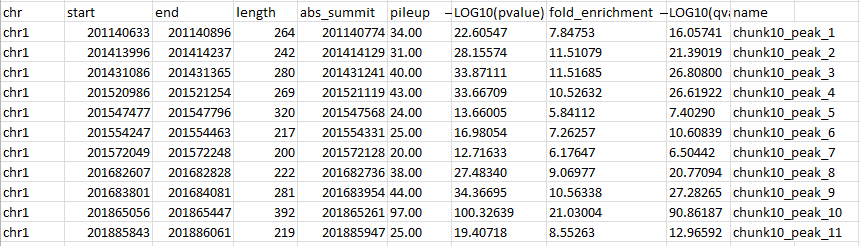
Как следует из результатов, были проанализированы чтения с первой хромосомы.
Всего было найдено 11 пиков.
Что касается характеристик пиков:

Наиболее достоверным пиком является пик 10, так как он обладает самым большим –log10p-value. Наименее достоверным является пик 7.
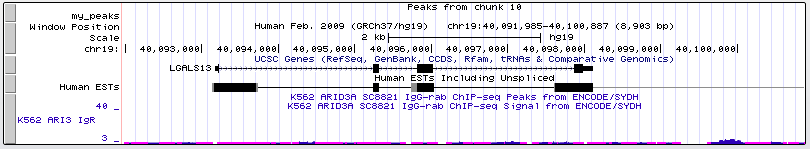
Информация была визуализирована из файла chunk10_peaks.narrowPeak с помощью геномного браузера UCSC Genome Browser
Для этого в файл chunk10_peaks.narrowPeak были добавлены строки
track type=narrowPeak visibility=3 db=hg19 name="my_peaks" description="Peaks from chunk 10" browser position chr1: 201140000- 201880000
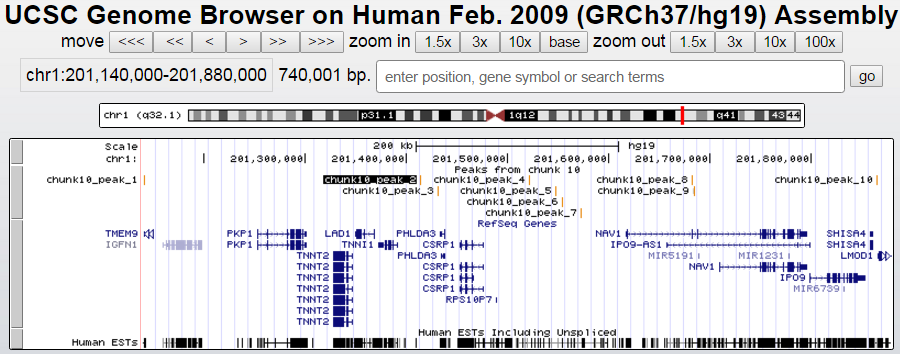
Пик 1 частично перекрывается с концом гена TMEM9

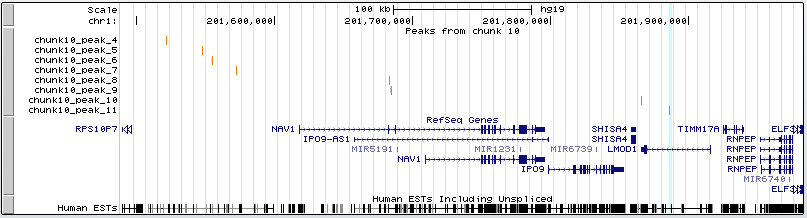
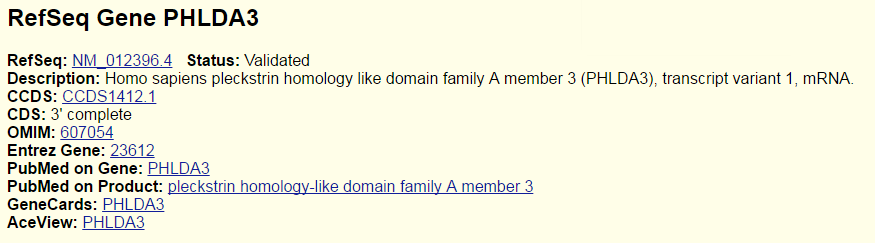
Как видно, пики 2-7 не лежат в области генов, однако пик 3 расположен недалеко от начала гена PHLDA3
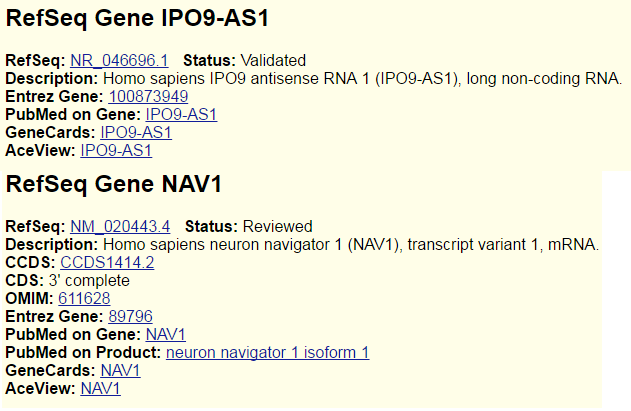
Пики 8 и 9 перекрываются с NAV1 и IPO9-AS1
Немного информации об этих генах:
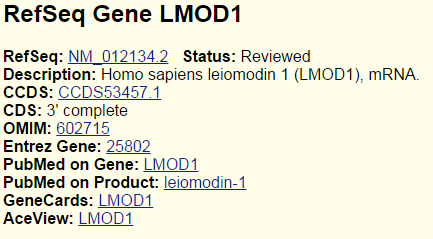
Пик 10 располагается в начале гена LMOD1, пик 11 также перекрывается с этим геном.
Задание 4. В геноме человека найдите три гена, транскрипция которых инициируется с помощью TATA-бокс связывающего белка, и один - без сигнала TATA-бокса в промоторной области
TATA-бокс связывающий фактор TBP - архейный и эукариотический белок, узнающий восьминуклеотидный сигнал в ДНК с консенсусом TATAWAAR(Faiger et al., 2005).Он является одним из ключевых ДНК-узнающих белков при образовании на промоторе генов комплекса TFIID инициации транскрипции с помощью Pol II (Lauder et al., 2016). Тем не менее, лишь часть промоторов имеет сигнал TATA-box, связываемый TBP.
Для поиска ТАТА-бокс был использован UCSC GenomeBrowser. И так: для анализа был выбран TPB и один из экспериментов.
Параметры выбранного эксперимента:

1. Ген, для которого был найден ТАТА-бокс (промоторная область в увеличенном масштабе):
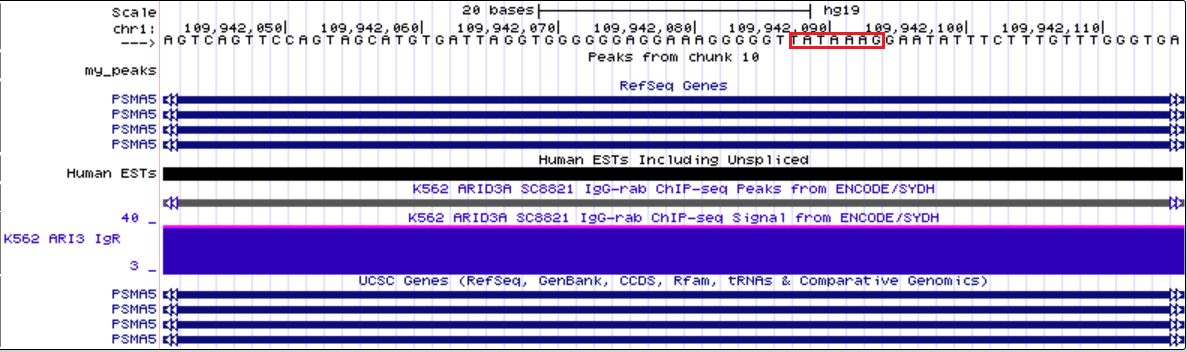
Промоторная область в уменьшенном масштабе
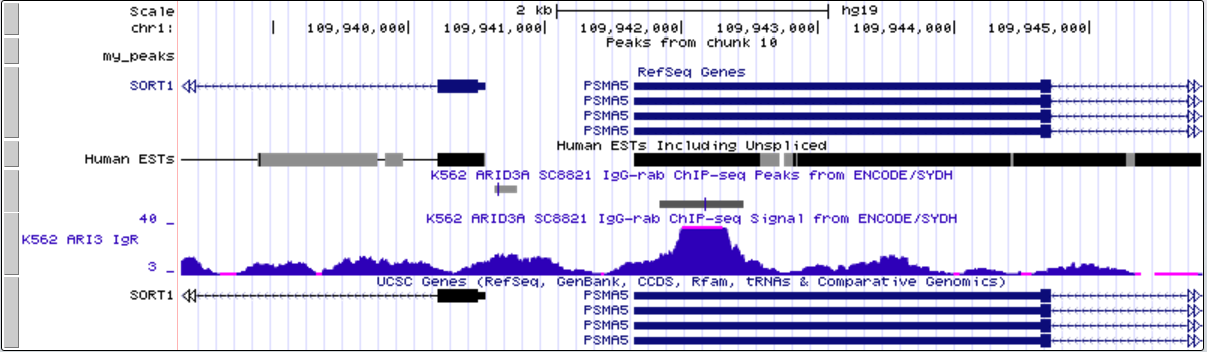
Ген PSMA5
Информация о гене: The proteasome is a multicatalytic proteinase complex with a highly ordered ring-shaped 20S core structure. The core structure is composed of 4 rings of 28 non-identical subunits; 2 rings are composed of 7 alpha subunits and 2 rings are composed of 7 beta subunits. Proteasomes are distributed throughout eukaryotic cells at a high concentration and cleave peptides in an ATP/ubiquitin-dependent process in a non-lysosomal pathway. An essential function of a modified proteasome, the immunoproteasome, is the processing of class I MHC peptides. This gene encodes a member of the peptidase T1A family, that is a 20S core alpha subunit. Multiple alternatively spliced transcript variants encoding two distinct isoforms have been found for this gene.

Координата старта транскрипции: 109 941 653
Координата ТАТА-бокса: 109 942 088
Последовательность ТАТА-бокса: ТАТАААG
2. Ген, для которого был найден ТАТА-бокс (промоторная область в увеличенном масштабе):
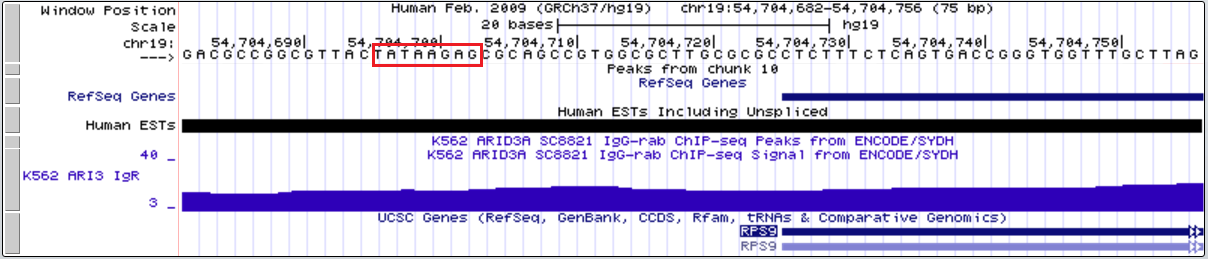
Промоторная область в уменьшенном масштабе:
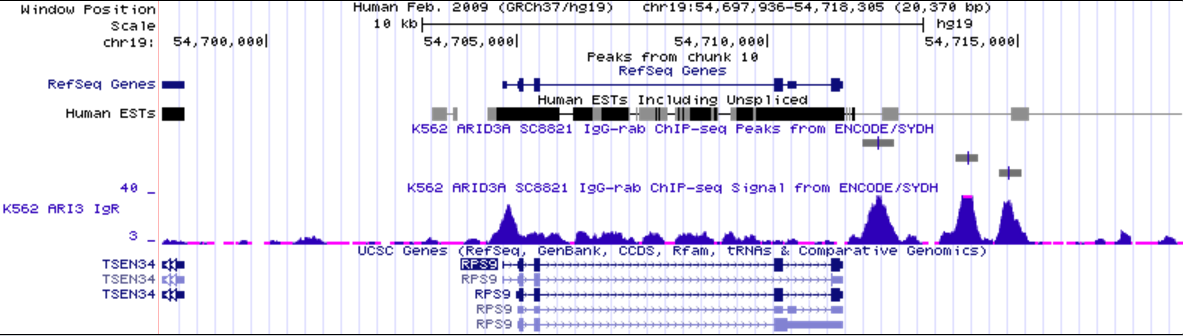
Ген RPS9
Информация о гене: Homo sapiens ribosomal protein S9 (RPS9), mRNA.

Координата старта транскрипции: 54 704 725
Координата ТАТА-бокса: 54 704 695
Последовательность ТАТА-бокса: ТАТААGAG
3. Ген, для которого был найден ТАТА-бокс (промоторная область в увеличенном масштабе):
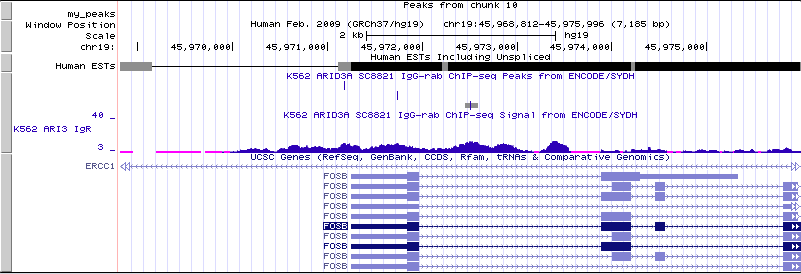
Промоторная область в уменьшенном масштабе:
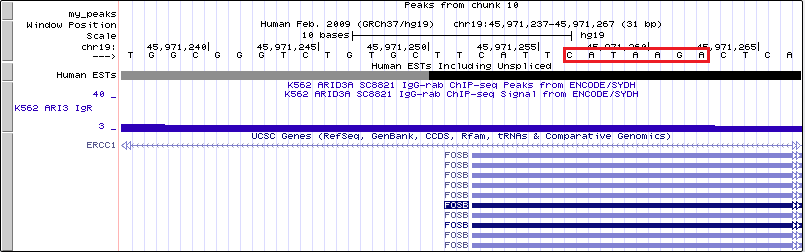
Ген FOSB
Информация о гене: Homo sapiens FBJ murine osteosarcoma viral oncogene homolog B (FOSB), transcript variant 1, mRNA.

Координата старта транскрипции: 45 971 253
Координата ТАТА-бокса:45 971 257
Последовательность ТАТА-бокса: САТААGA
4. Пример гена без ТАТА-бокса
Ген
Пиков в промоторной области ( и области за 400 нуклеотидов до промотора) нет, в отличии от генов с ТАТА-боксом, которые или имеют пики в промоторной области (как ген PSMA5), или в области прилежащей к промотору (гены FOSB и RPS9) .