Занятие 6: Программы парного выравнивания
Подсчёт веса выравнивания
Задание предполагает использование
выравнивая, полученного на предыдущем занятии, и матрицы
сходства BLOSUM62.
Штраф за открытие пробела - 12.
Штраф за удлинение пробела - 2.

| seq1 | - | A | V | A | A | D | T | P | G | P | V | S | P | C | G | A | C | R | Q | V | I | S |
| seq2 | I | A | V | V | A | D | T | P | V | P | - | D | P | C | G | A | C | R | Q | V | - | - |
| Вес | -12 | 4 | 4 | 0 | 4 | 6 | 5 | 7 | -3 | 7 | -12 | 0 | 7 | 9 | 6 | 4 | 9 | 5 | 5 | 4 | -12 | -2 |
Рассчитаем вес построенного нами выравнивания:
-12 + 4 + 4 + 0 + 4 + 6 + 5 + 7 + (-3) + 7 + (-12) + 0 + 7 + 9 + 6 + 4 + 9 + 5 + 5 + 4 + (-12) + (-2) = 45.
Построение выравнивания с помощью программы stretcher
stretcher является одной из программ пакета EMBOSS, выдающих парное выравнивание. Для выпонения задания запишем команду
stretcher seq1.fasta seq2.fasta stretcheralignment.stretcher -auto
Использованные файлы с последовательностями:
seq1.fasta,
seq2.fasta.
Полученный файл:
stretcheralignment.stretcher.
Выравнивание, построенное программой stretcher :
10 20 seq1 -AVAADTPGPVSPCGACRQVIS :: :::: : :::::::: seq2 IAVVADTPVP-DPCGACRQV-- 10
Сравнивание полученное выравнивание с построенным ранее, делаем вывод, что выравнивания совпадают.
Из информации, указанной в файле узнаём, что вес выравнивания равен 45. Как мы видим, посчитанный ранее вес выравнивания совпал с весом выравнивания, выданным программой.
Построение выравниваний с помощью программ needle и water
Программа needle пакета EMBOSS выдаёт оптимальное полное выравнивание.
Для построения полного выравнивания последовательностей белка CDD_BACSU (AC P19079) и родственного ему белка CDD_BACPY (AC Q9S3M0) с параметрами по умолчанию выполним следующую команду:
needle sw:p19079 sw:q9s3m0 needlealignment.needle -auto
В результате получим файл needlealignment.needle.
Полученное программой needle полное выравнивание:
CDD_BACSU 1 MNRQELITEALKARDMAYAPYSKFQVGAALLTKDGKVYRGCNIENAAYSM 50 |:.::||.|:.|||:.||.|||||.||||||.:||.:|.||||||:|||| CDD_BACPY 1 MDVEKLIAESKKAREQAYVPYSKFPVGAALLAEDGTIYHGCNIENSAYSM 50 CDD_BACSU 51 CNCAERTALFKAVSEGDTEFQMLAVAADTPGPVSPCGACRQVISELCTKD 100 .|||||||.|||||:|...|:.|||.|||.|||||||||||||:|.|... CDD_BACPY 51 TNCAERTAFFKAVSDGVRSFKALAVVADTEGPVSPCGACRQVIAEFCNGS 100 CDD_BACSU 101 VIVVLTNLQGQIKEMTVEELLPGAFSSEDLHDERKL 136 :.|.||||:|.|:|.||.:|||||||.|||....:. CDD_BACPY 101 MPVYLTNLKGDIEETTVAKLLPGAFSKEDLSYAAEQ 136
Для построения частичного выравнивания нам понадобится использовать программу water пакета EMBOSS. Выполним команду:
water sw:p19079 sw:q9s3m0 wateralignment.water -auto
В результате получим файл wateralignment.water.
Полученное программой water частичное выравнивание:
CDD_BACSU 1 MNRQELITEALKARDMAYAPYSKFQVGAALLTKDGKVYRGCNIENAAYSM 50 |:.::||.|:.|||:.||.|||||.||||||.:||.:|.||||||:|||| CDD_BACPY 1 MDVEKLIAESKKAREQAYVPYSKFPVGAALLAEDGTIYHGCNIENSAYSM 50 CDD_BACSU 51 CNCAERTALFKAVSEGDTEFQMLAVAADTPGPVSPCGACRQVISELCTKD 100 .|||||||.|||||:|...|:.|||.|||.|||||||||||||:|.|... CDD_BACPY 51 TNCAERTAFFKAVSDGVRSFKALAVVADTEGPVSPCGACRQVIAEFCNGS 100 CDD_BACSU 101 VIVVLTNLQGQIKEMTVEELLPGAFSSEDL 130 :.|.||||:|.|:|.||.:|||||||.||| CDD_BACPY 101 MPVYLTNLKGDIEETTVAKLLPGAFSKEDL 130
Подведём итоги:
- В частичное выравнивание вошёл участок последовательности от 1 до 130 «буквы» включительно.
- Локальное выравнивание совпадает с "ограничением" глобального на этот участок.
- Больший вес имеет локальное выравнивание (456.0 против 449.0 у глобального). Вообще, вес оптимального глобального выравнивания не может быть больше веса оптимального локального выравнивания из-за наличия в первом «невыгодных» с точки зрения наибольшего веса выравнивания участков.
Построение карты локального сходства с помощью программы dotmatcher
Программа dotmatcher пакета EMBOSS - программа с графическим выводом. Для построения карты воспользуемся последовательностями, использованными на предыдущем занятии. Выполним следующую команду:
dotmatcher seq1.fasta seq2.fasta
Ответив ps на вопрос программы о типе графика, мы получаем сообщение об успешном выполнении нашего запроса:
Created dotmatcher.ps
Полученный файл можно открыть программой GhostView:
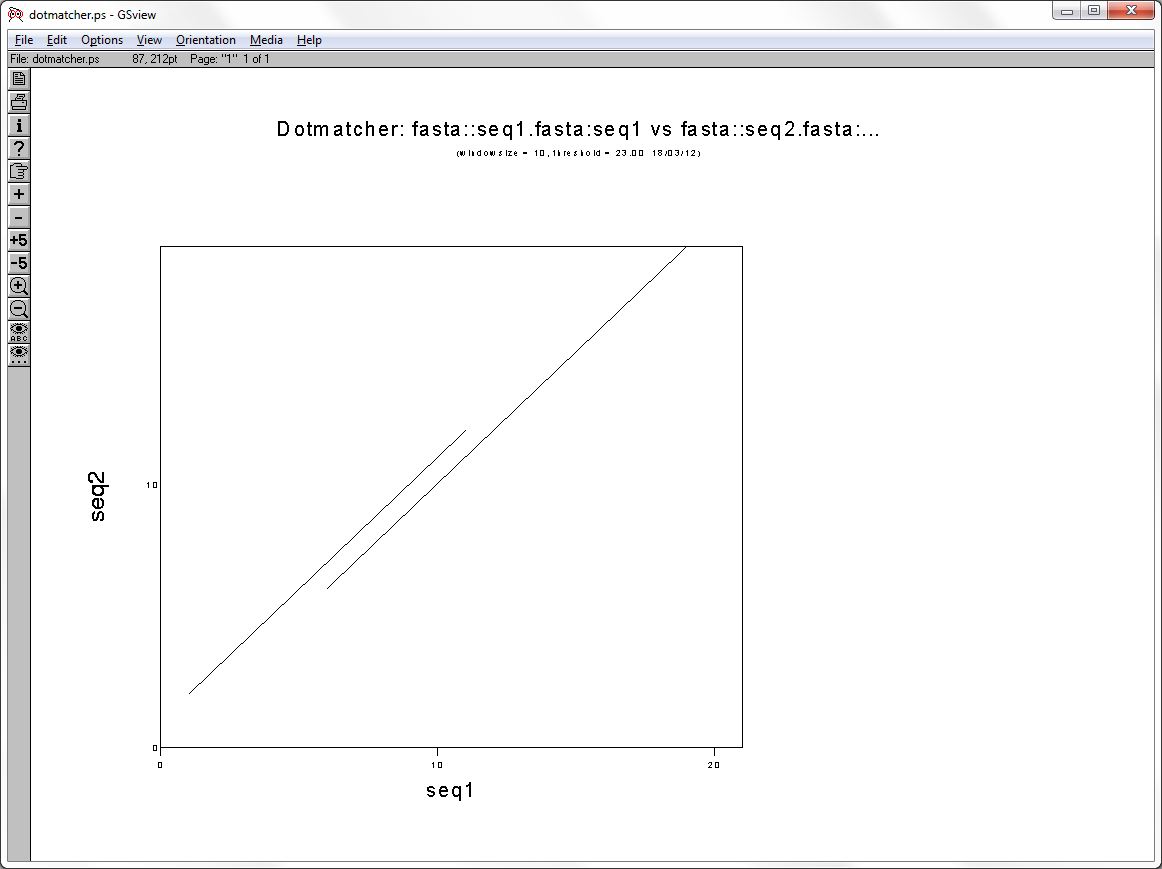
Файл dotmatcher.ps, открытый в программе GhostView
Построение субоптимальных локальных выравниваний с помощью программы matcher
C помощью программы matcher пакета EMBOSS можно получить несколько частичных выравниваний с наибольшим весом.
Выполним следующую команду (используем параметр -alternatives для указания количества альтернативных выравниваний, т.е. в файл будут сохранены дополнительные совпадения):
matcher seq1.fasta seq2.fasta matcheralignment2.matcher -alternatives 3
В результате получаем файл matcheralignment2.matcher. (Можно также сравнить его с файлом matcheralignment.matcher, полученным при всех параметрах по умолчанию: в последнем приведено только одно выравнивание с наибольшим весом, тогда как в matcheralignment2.matcher представлены 3 локальных выравнивания, как мы и указали в запросе.)
Полученные программой matcher локальные выравнивания:
10 seq1 AVAADTPGPVSPCGACRQV :: :::: : :::::::: seq2 AVVADTPVP-DPCGACRQV 10
Для первого выравнивания вес равен 69.
10 seq1 PGPVSPC : : : seq2 PDPCGAC 10
Для второго вес равен 20.
10 seq1 VAADTPGPV .: :: seq2 IAVVADTPV 9
Для третьего вес равен 12.
Как мы видим, полученные нами субоптимальные локальные выравнивания (второе и тертье выравнивания выше) действительно намного менее оптимальны, по сравнению с оптимальным локальным выравниванием (первое выравнивание).
Карта локального сходства последовательностей белков CDD_BACSU и CDD_BACPY
Аналогично приведённому выше заданию, воспользуемся программой dotmatcher пакета EMBOSS. Используем последовательности белка CDD_BACSU (AC P19079) и родственного ему белка CDD_BACPY (AC Q9S3M0). Выполним следующую команду:
dotmatcher sw:p19079 sw:q9s3m0
В результате получим файл, который можно открыть программой GhostView:
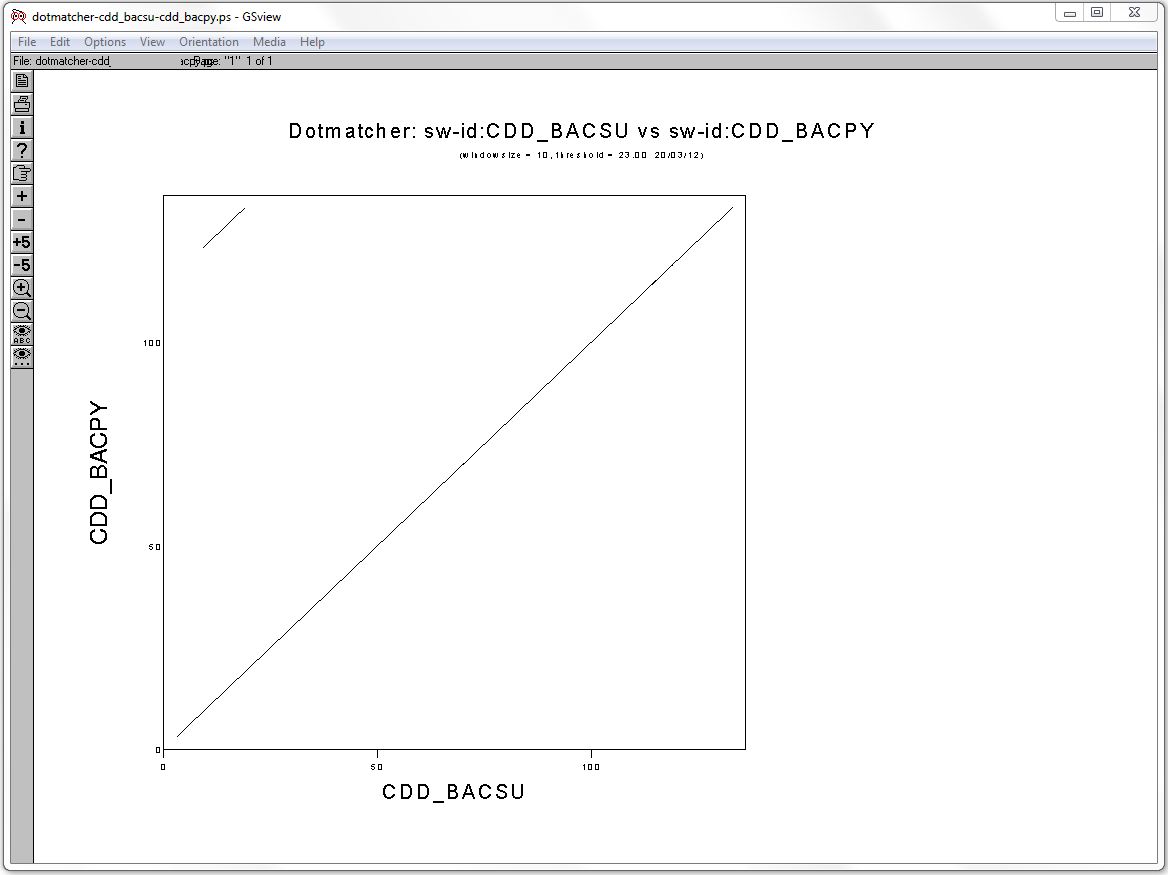
Файл, открытый в программе GhostView
Построение субоптимальных локальных выравниваний последовательностей белков CDD_BACSU и CDD_BACPY
C помощью программы matcher пакета EMBOSS получим несколько частичных выравниваний с наибольшим весом.
Выполним следующую команду:
matcher sw:p19079 sw:q9s3m0 matcheralignment-cdd_bacsu-cdd_bacpy.matcher -alternatives 3
В результате получаем файл matcheralignment-cdd_bacsu-cdd_bacpy.matcher.
Полученные программой matcher локальные выравнивания:
10 20 30 40 50 CDD_BA MNRQELITEALKARDMAYAPYSKFQVGAALLTKDGKVYRGCNIENAAYSM :. ..:: :. :::. :: ::::: :::::: .:: .: ::::::.:::: CDD_BA MDVEKLIAESKKAREQAYVPYSKFPVGAALLAEDGTIYHGCNIENSAYSM 10 20 30 40 50 60 70 80 90 100 CDD_BA CNCAERTALFKAVSEGDTEFQMLAVAADTPGPVSPCGACRQVISELCTKD ::::::: :::::.: :. ::: ::: :::::::::::::.: : CDD_BA TNCAERTAFFKAVSDGVRSFKALAVVADTEGPVSPCGACRQVIAEFCNGS 60 70 80 90 100 110 120 130 CDD_BA VIVVLTNLQGQIKEMTVEELLPGAFSSEDL . : ::::.: :.: :: .::::::: ::: CDD_BA MPVYLTNLKGDIEETTVAKLLPGAFSKEDL 110 120 130
Для первого выравнивания вес равен 456.
10 CDD_BA ELITEALKARDMAYA .:. : :..:: CDD_BA KLLPGAFSKEDLSYA 120 130
Для второго вес равен 27.
120 CDD_BA MTVEELL : ::.:. CDD_BA MDVEKLI 7
Для третьего вес равен 20.
Как и в предыдущем случае, делаем вывод, что, полученные нами субоптимальные локальные выравнивания (второе и тертье выравнивания выше) действительно намного менее оптимальны, по сравнению с оптимальным локальным выравниванием (первое выравнивание).
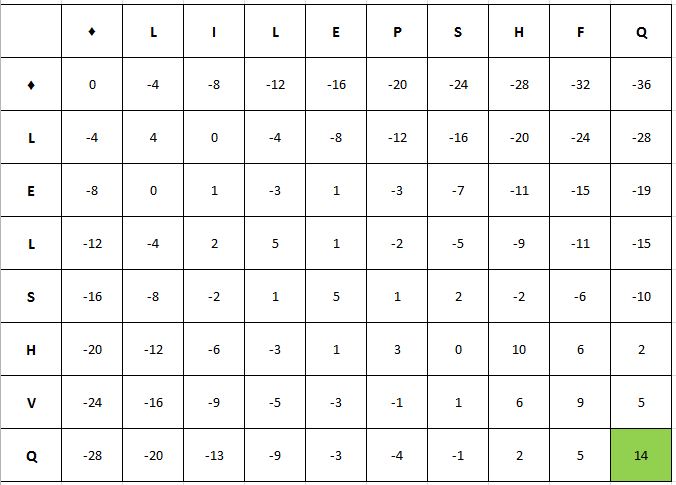
Поиск веса оптимального выравнивая последовательностей
Задание выполнено с использованием Excel. В качестве матрицы весов замен была использована матрица BLOSUM62. В файле представлена карта сходства двух последовательностей, а также карта оптимальных весов для последовательностей из файла handout.
Загрузить файл handout_optimal_weight.xlsx с результатами.
Пользуясь этой картой, мы можем определить вес оптимального выравнивания данных последовательностей: он равен 14.
Ссылки
- Файл seq1.fasta - последовательность первого фрагментов в fasta-формате.
- Файл seq2.fasta - последовательность второго фрагментов в fasta-формате.
- Файл stretcheralignment.stretcher выравнивание, полученное программой stretcher.
- Файл needlealignment.needle - выравнивание, полученное программой needle.
- Файл wateralignment.water - выравнивание, полученное программой water.
- Файл dotmatcher.ps - карта локального сходства, полученная программой dotmatcher.
- Файл matcheralignment2.matcher, содержащий субоптимальные локальные выравнивания, построенные программой matcher.
- Файл dotmatcher-cdd_bacsu-cdd_bacpy.ps - карта локального сходства для последовательностей белка и его гомолога, полученная программой dotmatcher.
- Файл matcheralignment-cdd_bacsu-cdd_bacpy.matcher, содержащий субоптимальные локальные выравнивания последовательностей белка и его гомолога, построенные программой matcher.
- Файл handout_optimal_weight.xlsx, содержащий карту сходства и карту оптимальных весов для двух последовательностей.