Занятие 5: Выравнивание последовательностей
Построение выравнивания фрагментов последовательности двух родственных белков вручную
Задание предполагает выполнение построения выравнивания в программе GeneDoc.

Импортировав в программу файл shortseqs.fasta, содержащий пару коротких последовательностей в fasta-формате, мы можем выровнять последовательности, стараясь, чтобы было сопоставлено максимальное число одинаковых букв:

Данное выравнивание сохранено в файле alignment1.msf.
Рассчитаем процент идентичности двух последовательностей:
число колонок с одинаковыми буквами - 15;
общее число колонок выравнивания - 22;
процент идентичности - (15 / 22) * 100% = 68,18%.
Пользуясь матрицей сходства BLOSUM62, рассчитаем процент сходства двух последовательностей:
число колонок с одинаковыми буквами - 15;
число колонок с буквами, соответствующими сходным остаткам, - 0;
общее число колонок выравнивания - 22;
процент идентичности - ((15 + 0) / 22) * 100% = 68,18%.
Построение карты локального сходства последовательностей с использованием возможностей Excel
Для введения последовательности по букве в ячейке можно воспользоваться функцией Текст по столбцам в меню Данные (и применить транспонирование текста для записи последовательности в столбец по букве в ячейке) или же написать формулу (см. файл alignment1_map.xlsx).
Для отметки совпадающих букв цифрой 1 можно также составить формулу (см. файл alignment1_map.xlsx).
Цветом закрашены ячейки, образующие путь, соответствующий оптимальному, на мой взгляд, выравниванию.
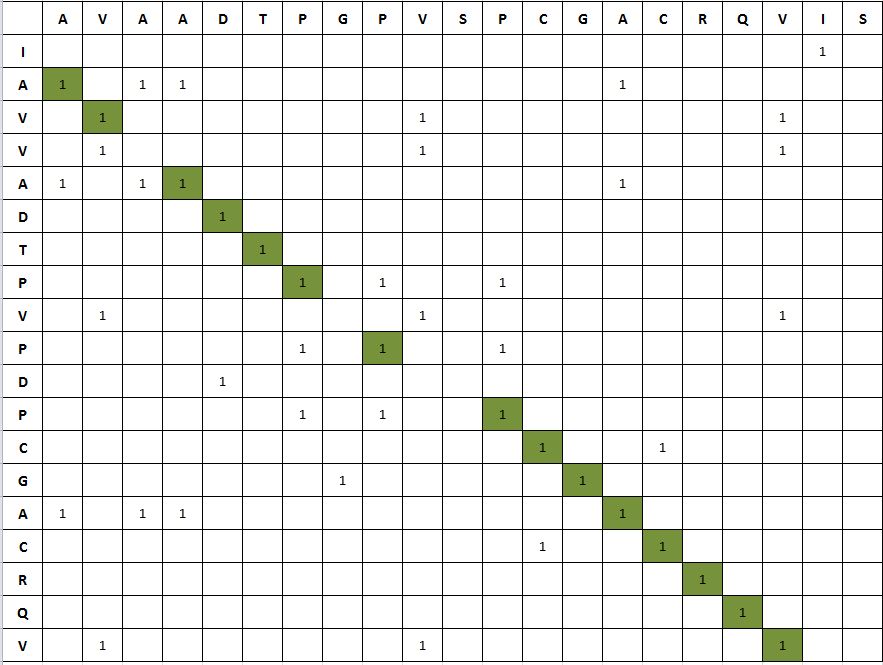
Мы можем проследить соответствие между путём на карте и выравниванием. Переход к следующей клетке по диагонали (i+1, j+1) (имеется в виду, что они обе закрашены)
соответствует идущим подряд одинаковым буквам в выравнивании последовательностей.
Как мы видим, переход к следующей через одну закрашенной клетке по диагонали (i+2, j+2) означает, что между одинаковыми буквами есть 1 буква в
каждой из последовательностей, при этом эти буквы не совпадают. Переход на 2 клетки вправо и на 1 клетку вниз (i+2, j+1) будет означать, что в последовательности, записанной
в столбец, при данном выравнивании есть один гэп. Аналогично, переход i+1, j+2 говорит о наличии гэпа в последовательности, записанной в строку.
На карте мы видим переход i+3, j+2, что означает, что в данном выравнивании между совпадающими буквами (их совпадение обозначено закрашиванием
клеток (i, j) и (i+3, j+2) на карте) присутствует 1 несовпадающая буква в каждой из последовательностей и 1 гэп в последовательности, записанной в столбец.
Выравнивание фрагмента с последовательностью белка CDD_BACSU с помощью программы bl2seq
Программа bl2seq строит частичные выравнивания. bl2seq стоит на kodomo и реализована как сервис на сайте NCBI BLAST.
На странице выравнивания аминокислотных последовательностей выполним необходимый нам запрос, задав AC белка (P19079) и искомый фрагмент последовательности.
В результате получаем следующие данные:
>lcl|50483 unnamed protein product Length=21 Score = 45.4 bits (106), Expect = 2e-13, Method: Composition-based stats. Identities = 21/21 (100%), Positives = 21/21 (100%), Gaps = 0/21 (0%) Query 74 AVAADTPGPVSPCGACRQVIS 94 AVAADTPGPVSPCGACRQVIS Sbjct 1 AVAADTPGPVSPCGACRQVIS 21
Соответственно, координаты последовательности фрагмента в полной последовательности белка: 74 - 94.
Выравнивание последовательности белка CDD_BACSU с последовательностью гомологичного белка CDD_BACPY
В задании предполагается использование сервиса bl2seq для построения частичного выравнивания последовательностей белков CDD_BACSU (AC P19079) и CDD_BACPY (AC Q9S3M0).
| ID | CDD_BACSU | CDD_BACPY |
| Организм | Bacillus subtilis (Сенная палочка) | Bacillus psychrophilus (Sporosarcina psychrophila) |
| Процент идентичности (Identities) | 68% | |
| Процент сходства (Positives) | 79% | |
| Число гэпов | 0 | 0 |
| Число идущих подряд гэпов | 0 | 0 |
| Суммарное число гэповых колонок | 0 | |
| Координаты выровненных участков | 1 - 130 | 1 - 130 |
Выравнивание, построенное сервисом bl2seq:
>sp|Q9S3M0.1|CDD_BACPY RecName: Full=Cytidine deaminase; Short=CDA; AltName: Full=Cytidine aminohydrolase emb|CAB51906.1| cytidine deaminase [Sporosarcina psychrophila] Length=136 Score = 185 bits (469), Expect = 4e-65, Method: Compositional matrix adjust. Identities = 88/130 (68%), Positives = 103/130 (79%), Gaps = 0/130 (0%) Query 1 MNRQELITEALKARDMAYAPYSKFQVGAALLTKDGKVYRGCNIENAAYSMCNCAERTALF 60 M+ ++LI E+ KAR+ AY PYSKF VGAALL +DG +Y GCNIEN+AYSM NCAERTA F Sbjct 1 MDVEKLIAESKKAREQAYVPYSKFPVGAALLAEDGTIYHGCNIENSAYSMTNCAERTAFF 60 Query 61 KAVSEGDTEFQMLAVAADTPGPVSPCGACRQVISELCTKDVIVVLTNLQGQIKEMTVEEL 120 KAVS+G F+ LAV ADT GPVSPCGACRQVI+E C + V LTNL+G I+E TV +L Sbjct 61 KAVSDGVRSFKALAVVADTEGPVSPCGACRQVIAEFCNGSMPVYLTNLKGDIEETTVAKL 120 Query 121 LPGAFSSEDL 130 LPGAFS EDL Sbjct 121 LPGAFSKEDL 130
Карта локального сходства (Dot Matrix View):
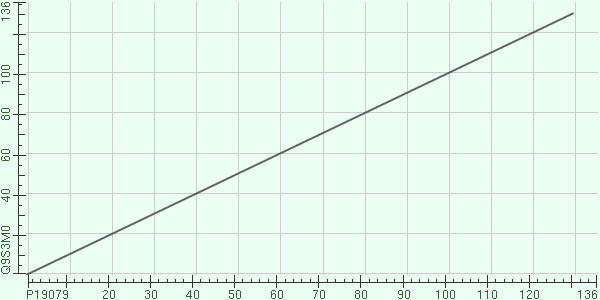
Загрузить карту локального сходства в формате .gif.
Создание матрицы, в ячейках которой стоит вес замены соответствующих остатков в строке и столбце
Задание выполнено с использованием Excel. В качестве матрицы весов замен была использована матрица BLOSUM62.
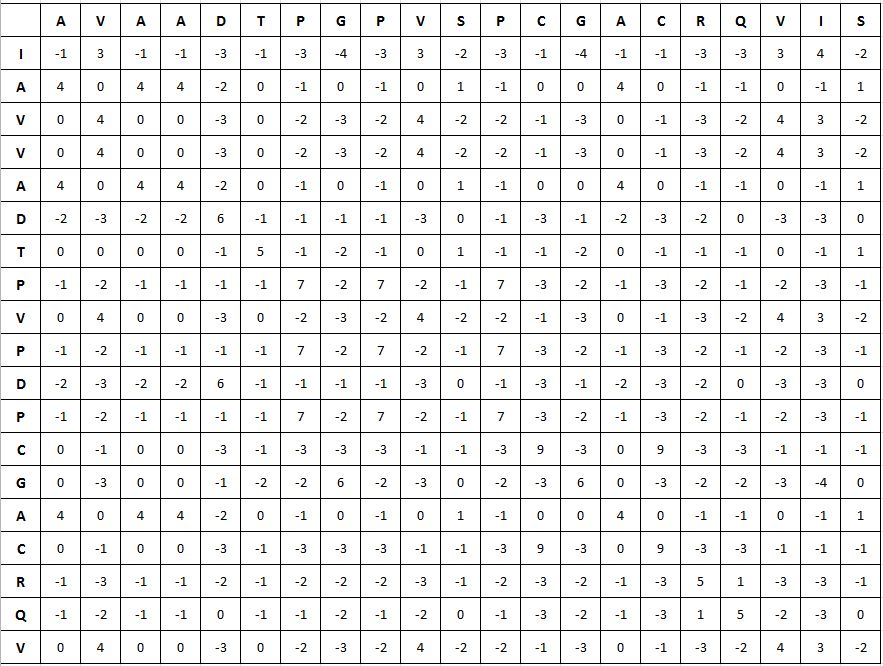
Загрузить файл alignment1_weight.xlsx.
Сравнение выравниваний последовательностей, построенных с разными параметрами программы bl2seq
Если изменить матрицу с BLOSUM62 на PAM70, то процент идентичности не изменится (что естественно), а процент сходства уменьшится с 79% до 75%.
Для характеристики того, насколько одинаковы два выравнивания (одних и тех же последовательностей), можно использовать процент согласованных столбцов двух выравниваний относительно числа столбцов в каждом из выравниваний. В данном случае при изменении матрицы процент согласованных столбцов равен 100%.
Если изменять штрафы за гэпы, то мы замечаем, что процент согласованных столбцов двух выравниваний равен 100%. Это неудивительно, ведь в рассматриваемом случае в последовательностях при выравнивании вообще отсутствуют гэпы.
Воспроизведение выравнивания, полученного программой bl2seq, в GeneDoc
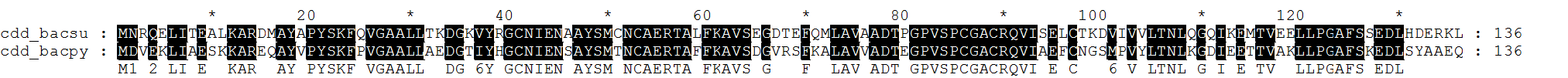
> Изображение в оригинальном размере.
> Загрузить файл (1) с выравниванием.
Импортировав последовательности белков CDD_BACSU и CDD_BACPY в GeneDoc, мы можем сделать полное выравнивание.
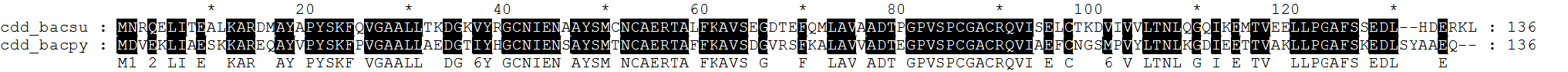
> Изображение в оригинальном размере.
> Загрузить файл (2) с выравниванием.
P.S. Отличие между двумя последними выравниями, приведёнными выше, небольшое - во втором случае добавлено 2 гэпа в первую последовательность для совпадения в двух последовательностях остатка E (глутаминовой кислоты).
Ссылки
- Файл shortseqs.fasta - последовательности двух фрагментов в fasta-формате.
- Файл alignment1.msf - выравнивание двух фрагментов последовательностей.
- Файл alignment1_map.xlsx, содержащий карту локального сходства для двух фрагментов последовательностей.
- Файл cdd_bacsu_&_cdd_bacpy.gif - карта локального сходства для последовательностей белков CDD_BACSU и CDD_BACPY.
- Файл alignment1_weight.xlsx, содержащий матрицу с весом замены соответствующих остатков в ячейках для двух фрагментов последовательностей.
- Файл cdd_bacsu_&_cdd_bacpy_2.msf - полное выравнивание последовательностей белков CDD_BACSU и CDD_BACPY.