Занятие 8: Множественное выравнивание последовательностей
Выравнивание набора гомологов белка CDD_BACSU
C помощью BLAST на NCBI составим выборку из нескольких гомологов белка CDD_BACSU (AC P19079), учитывая при этом следующие моменты:
- E-value < 0.001 (можно установить такой порог при поиске);
- процент идентичности выравнивания - от 40% до 80%;
- отсутствие одинаковых белков из слишком родственных организмов;
- желательно ограничение выдачи таксоном Bacteria.
Полученную выборку можно представить в виде файла myproteins.list, содержащего адреса последовательностей:
sw:cdd_bacpy
sw:cdd_mycpi
sw:cdd_human
sw:cdd_mouse
sw:cdd_dicdi
sw:cdd_schpo
sw:cdd_yeast
Чтобы получить последовательности в fasta-формате, выполним следующую команду (знак @ указывает программе, что входной файл следует рассматривать как лист-файл):
seqret @myproteins.list myproteins.fasta
В результате получим файл myproteins.fasta, содержащий указанные выше последовательности в fasta-формате.
Пользуясь возможностями программы JalView построим множественное выравнивание отобранных последовательностей, например, с помощью Clustral:

Изображение (1) в оригинальном размере.

Изображение (2) в оригинальном размере.
В структуре выравнивания можно выделить некоторые особенности:
- Наблюдаются участки с повышенной долей консервативных позиций.
Координаты этих участков по столбцам выравнивания: 37 - 39, 58 - 66 (в частности, наиболее консервативны 58, 60, 62 - 63 и 66 столбцы), 71 - 74, 104 - 111 (в частности, 105 - 107 и 109 - 111), 141 - 143 (к тому же, имеются "одиночные" консервативные позиции: 41, 66, 76, 84, 115, 146 и 151 столбцы).
Участки с повышенной долей консервативных позиций в координатах по остаткам белка CDD_BACSU:
20 - 22, 40 - 48, 53 - 56, 84 - 91, 120 - 122.
(Если производить выделение участков с меньшей точностью, то можно выделить 4 основных участка с повышенной долей консервативных позиций, которые (участки) хорошо различимы на изображении множественного выравнивания: 34 - 48, 58 - 84, 104 - 115, 141 - 146 в координатах по столбцам выравнивания.) - С определённой долей вероятности можно утверждать, что на участках 1 - 17 и 156 - 166 в координатах по столбцам выравнивания оно не имеет биологического смысла.
- Функционально консервативные позиции в представленном выравнивании образуют группы (в скобках указано число таких групп в выравнивании) QE (2), TS (2), VI (4), FY (2), ML (2), LVIM (2), RK (2), FW (1), LIM (3), DN (1). Наибольшее число раз среди функционально консервативных позиций встречается группа VI, а также группа LIM.
- Если к группам сходных аминокислот добавить DE (аспартат и глутамат), то можно обозначить консервативную позицию по столбцу 21 (4 остаток в белке CDD_BACSU), а также по столбцу 149 (128 остаток в белке CDD_BACSU).
Загрузить файл с выравниванием в формате .msf.
Дополнение в связи с изменением задания
Нахождение функционально консервативных позиций наиболее удобно при рассмотрении выравнивания с раскраской по консервативности (при этом порог консервативности ≈50) с использованием схемы раскраски BLOSUM62, что демонстрирует следующее изображение:

Изображение (3) в оригинальном размере.
Функционально консервативные позиции в выравнивании образуют следующие группы сходных аминокислот (в скобках указано число таких групп в выравнивании): AS (1), VI (1), LIV (3), LV (1), IV (2), ST (2).
Таким образом, среди функционально консервативных позиций наибольшее число раз встретилась группа LIV, которая встречается в выравнивании 3 раза. Второе место по частоте встречаемости разделяют группы IV и ST - они встречаются в выравнивании по 2 раза.
Загрузить файл с выравниванием в формате .jar.
Работа с программой Muscle
С помощью SRS осуществим поиск последовательностей малых дельта-антигенов в банке Swiss-Prot:

Сохранив полученные результаты поиска, получим файл delta.fasta, содержащий последовательности малых дельта-антигенов в fasta-формате.
Выровнять последовательности можно программой muscle. Для этого выполним следующую команду:
muscle -in delta.fasta -out delta_aligned.fasta
В результате получим файл delta_aligned.fasta, содержащий выравнивание последовательностей из файла delta.fasta.
Получить аналогичное выравнивание программой Muscle можно также в программе JalView:
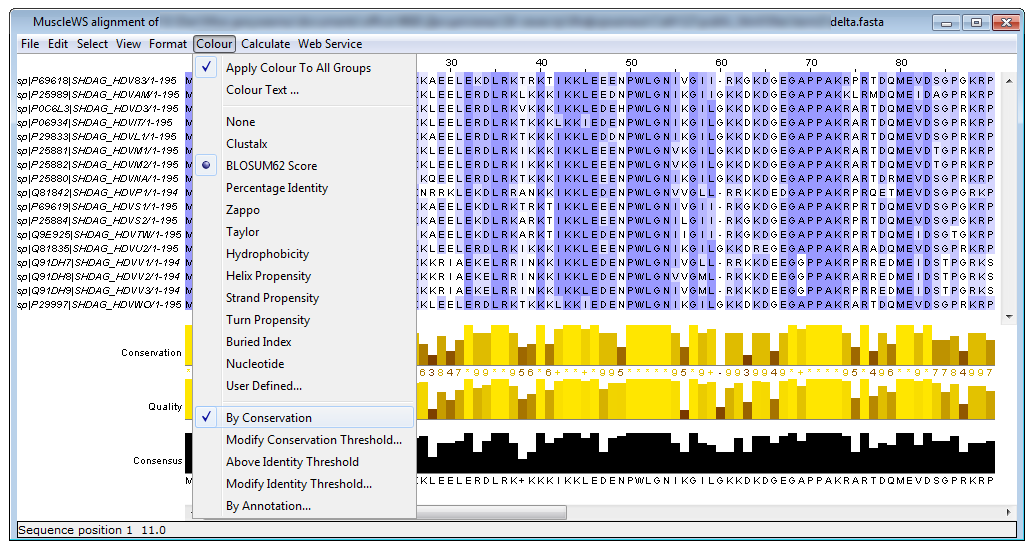
Пользуясь возможностями JalView, выберем необходимый тип окраски позиций (BLOSUM62 - по функциональным группам остатков, т.е. с учётом матрицы весов замен, By Conservation - по степени консервативности):
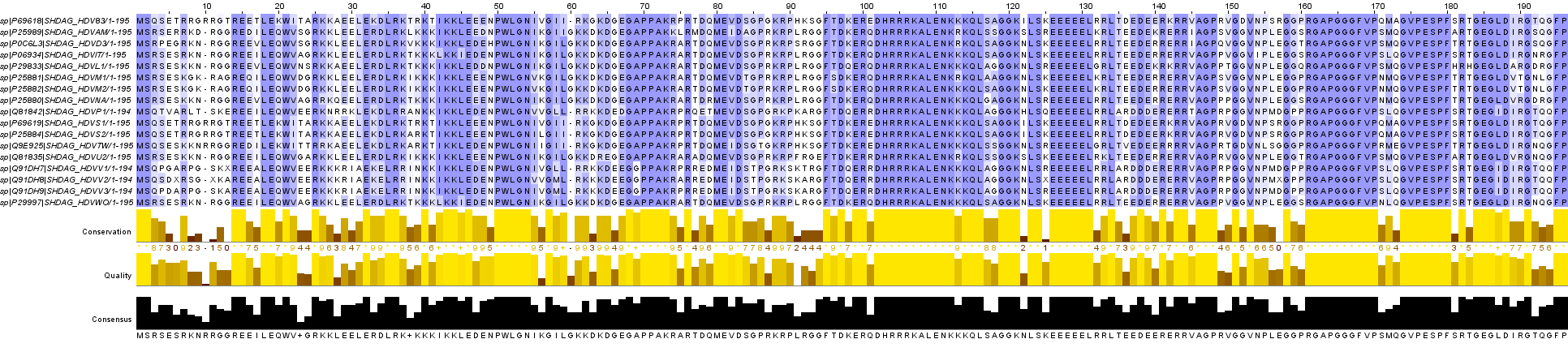
Рассмотрим подробнее изображение выравнивания последовательностей малых дельта-антигенов, полученного с помощью программы JalView (File → Export Image → PNG):
Изображение в оригинальном размере.
Непосредственно под выровненными последовательностями малых дельта-антигенов расположены
Conservation наглядно изображает оценку консервативности соответствующей колонки. Conservation измеряется как
числовой индекс, отражающий консервативность (сохранение) физико-химических свойств выравнивания: учитываются вес идентичности (Identities score) и
замены аминокислот из тех же физико-химических классов. Соответственно, цифры под гистограммой, отражающей консервативность,
соответствуют указанному индексу; знак
+ соответствует индексу 10.0, * - индексу 11.0.
Цвета колонок гистограммы и соответствующих им цифр также зависят от индекса: от тёмно-коричневого до ярко-жёлтого по мере увеличения индекса.
Quality показывает в виде гистограммы качество колонки, зависящее от суммы весов замен по матрице BLOSUM62.
Consensus наглядно изображает наиболее часто встречаемую букву в соответствующей колонке выравнивания. При наведении
курсора мыши на столбец гистограммы можно узнать процент встречаемости наиболее частой буквы в соответствующей колонке выравнивания.
Если несколько разных букв встречаются в колонке одинаковое число раз, то под соответствующим столбцом гистограммы ставится знак +,
а при наведении курсора мыши на этот столбец отображаются наиболее частые буквы с указанием процента встречаемости.
Загрузить файл с выравниванием в формате .msf.
Загрузить файл с выравниванием в формате .jar.
Формат jalview (.jar) позволяет сохранять выравнивания с дополнительной информацией, такой как окраска, аннотации и деревья ("whole sets of coloured and annotated alignments and trees can be read from").
Отображение консервативных участков последовательности на структуре
Используя средства JalView, свяжем последовательность белка CDD_BACSU с файлом PDB (в задании используется выравнивание, полученное при изучении последовательностей гомологов белка CDD_BACSU в первом задании текущего занятия):
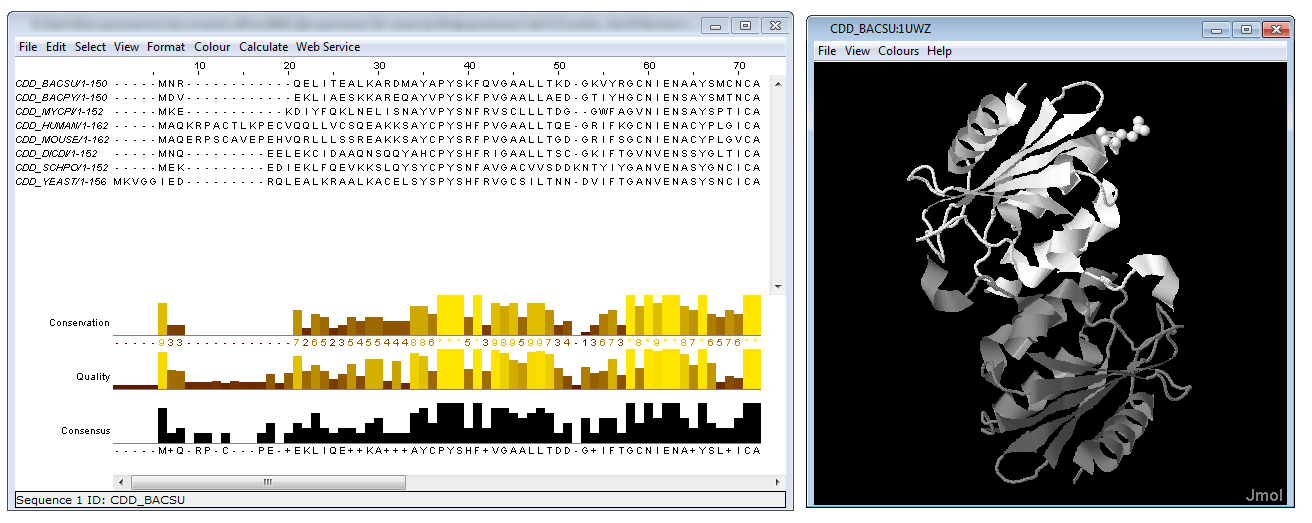
Затем раскрасим выравнивание по консервативности, используя схему раскраски BLOSUM62:
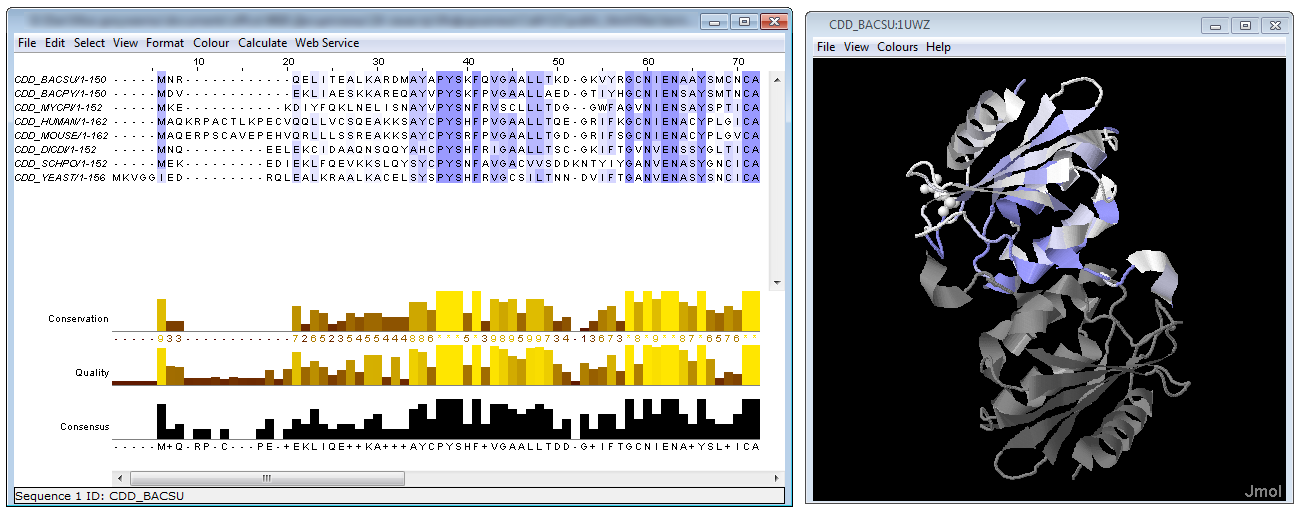
Заметим, что раскраска последовательности отображается на структуре (но не наоборот).
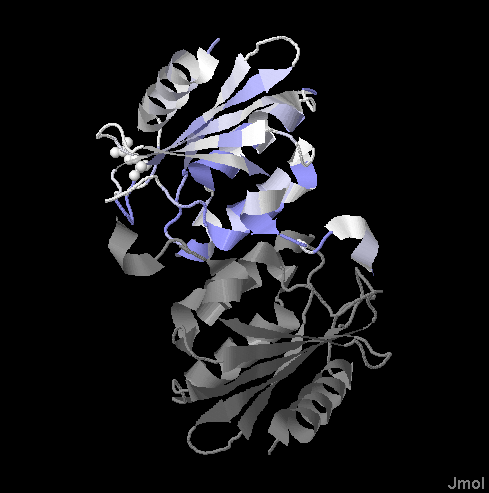
Значительная часть консервативных остатков расположена на поверхности глобулы. Они также присутствуют в ядре глобулы. Консервативные участки наблюдаются как в петлях, так и в элементах вторичной структуры (в α-спиралях и β-листах). Остатки, связывающие лиганд, относительно неконсервативны (значение индекса консервативности - 5).
Загрузить файл с выравниванием в формате .jar.
Другие программы множественного выравнивания
mafft
Информацию об использовании программы множественного выравнивания mafft можно получить, например, выполнив команду
mafft -help
Программа mafft имеет несколько вариантов (опций) исполоьзования: построение выравнивания с высокой скоростью, с высокой точностью, а также автоматический выбор опции (на тот случай, когда мы не уверены, какую опцию стоит использовать в конкретном случае).
Среди параметров программы mafft следует выделить следующие:
- --op - штраф за открытие гэпа (по умолчанию равен 1.53);
- --ep - компенсация (фактически, штраф за продление гэпа; по умолчанию 0.0);
- --maxiterate - максимальное число повторных улучшений (по умолчанию 0.0);
- --clustalout - выходной формат файла выравнивания (по умолчанию .fasta);
- --reorder - порядок выровненных последовательностей в выходном файле (по умолчанию в том порядке, в котором они были введены);
- --quiet - отключение сообщений о текущем прогрессе процесса выравнивания;
- --thread - число потоков; оно не должно быть больше числа физических ядер.
Используем программу mafft, например, для построения выравнивания последовательностей белка CDD_BACSU и его гомологов. Для этого воспользуемся файлом myproteins.fasta и выполним следующую команду:
mafft --auto myproteins.fasta > mafft_alignment.fasta
Программа mafft автоматически выбрала медленный, но самый точный алгоритм для построения выравнивания. В результате мы получили файл mafft_alignment.fasta.
edialign
Описание программы edialign можно найти в разделе пакет EMBOSS страницы с описаниями программ:
edialignС помощью программы edialign построим выравнивание последовательностей, записанных в файле myproteins.fasta:
edialign myproteins.fasta myproteins.edialign editalign_alignment.fasta
В результате мы получим файл edialign_alignment.fasta,содержащий выравнивание последовательностей и файл myproteins.edialign, в котором записана информация о выравнивани.
Сравнивая полученные файлы, мы можем сделать вывод о несовпадении выравниваний (впрочем, имеется большое количество сходных выровненных участков). Причиной этому может быть различие в алгоритмах построения выравнивания, а также различие в используемых параметрах по умолчанию.
Изображение послученных выравниваний, открытых в программе JalView:
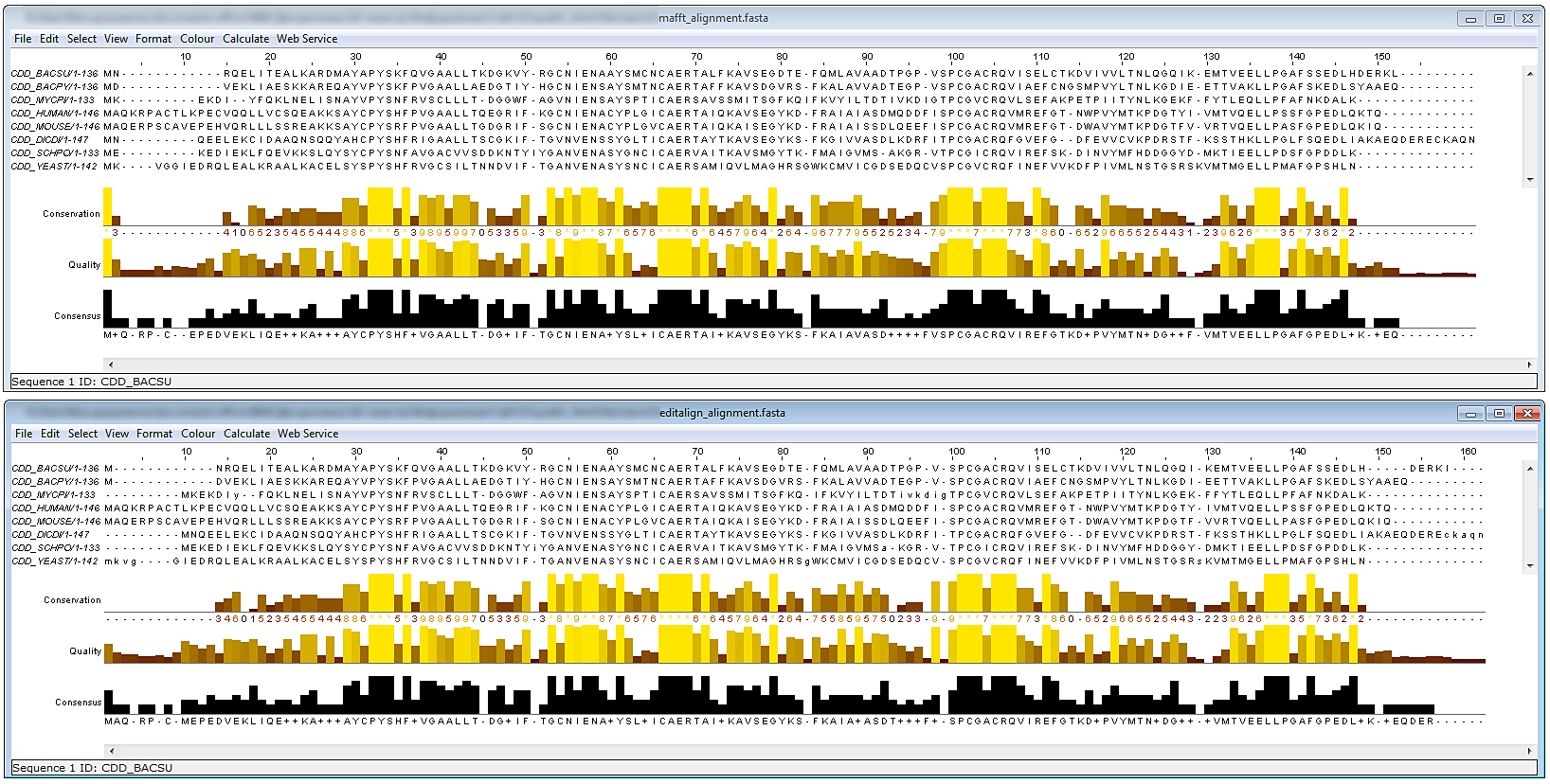
Изображение в оригинальном размере.
Знакомство с некоторыми программами обработки множественных выравниваний
Раздел пакет EMBOSS страницы с описаниями программ дополнен описаниями следующих программ:
consambig distmat plotconНекоторые возможности программы JalView
Краткая информация о некоторых возможностях программы JalView представлена на соответствующей странице.
Ссылки
- Файл myproteins.list, содержащий идентификаторы последовательностей белка CDD_BACSU и найденных гомологов.
- Файл myproteins.fasta, содержащий последовательности белка CDD_BACSU и найденных гомологов.
- Файл jalview_alignment1.msf, содержащий множественное выравнивание последовательностей белка CDD_BACSU и найденных гомологов.
- Файл delta.fasta, содержащий последовательности малых дельта-антигенов.
- Файл delta_aligned.fasta, содержащий множественное выравнивание последовательностей малых дельта-антигенов.
- Файл delta.msf, содержащий множественное выравнивание последовательностей малых дельта-антигенов, выполненное в программе JalView.
- Файл delta.jar, содержащий множественное выравнивание последовательностей малых дельта-антигенов, выполненное в программе JalView, с раскраской по консервативности с использованием схемы раскраски BLOSUM62.
- Файл jalview_alignment1_withstructure.jar, содержащий множественное выравнивание последовательностей белка CDD_BACSU и найденных гомологов с раскраской по консервативности с использованием схемы раскраски BLOSUM62, при этом последовательность белка CDD_BACSU связана с файлом структуры белка.