Занятие 1: Linux. Описание программ bash.
Раздел 1: bash

bash - Одна из наиболее популярных современных разновидностей командной оболочки UNIX; особенно популярна в среде Linux, где она часто используется в качестве предустановленной командной оболочки. источник
Стандартные потоки и работа с ними
Каждое консольное приложение имеет дело с тремя потоками: stdin, stdout, stderr.
stdin - стандратный поток ввода - содержит информацию, которую мы набираем на клавиатуре во время работы программы. Этот поток имеет номер 0.
stdout - стандратный поток вывода - зарезервирован для вывода данных. Это поток номер 1.
stderr - стандратный поток ошибок - зарезервирован для вывода диагностических и отладочных сообщений в текстовом виде. Это поток номер 2.
В консольном окне по умолчанию отображается сожержание потоков stdout и stderr.
Поток stdout можно перенаправить с помощью символа > в определенное пользователем место, например в файл.
Если требуется произвести запись в конец файла, то следует использовать символ >>.
Для вывода потока stderr используется символ 2>.
При работе с различными командами удобно использовать конвейер, который задаётся знаком | и применяется для направления stdout одной команды на вход (stdin) другой команды. Для направления в конвейер stderr используется выражение 2>&1|.
Рассмотрим несколько примеров.
ls | wc
Получив список содержимого текущей директории, мы передаём результат на вход команде wc, которая подсчитывает слова, строки и байты.
man find > find_manual.txt
Так можно записать информацию об использовании команды find в указанный файл.
Спецсимволы
Некоторые символы воспринимаются интерпретатором командной строки bash как указания произвести некоторые операции.
Пробел и символ табуляции отделяют друг от друга параметры.
Среди кавычек следует различать одинарные, двойные и обратные.
' - все символы в одинарных кавычках воспринимаются буквально; выражение объединяется, но не интерпретируется.
" - выражение в двойных кавычках объединяется и интерпретируется; символы $ и ` имеют специальное значение
(вызов содержания переменной и подстановка вывода другой команды); сочетание \ с любым символом превращается в этот символ.
` - выражение в обратных кавычках выполняется. Пример: можно выполнить команду, похожую на приведённую выше - wc `ls`.
Для поиска файлов часто приходится работать со своеобразным шаблоном - маской файла. Так, * заменяет собой любые символы или их отсутствие. ? заменяет какой-либо один символ. С помощью [ ] можно задавать диапазон цифр или букв, например, запись [0-9] обозначает одну цифру.
Символ ! можно использовать для вызова команды из журанала команд. Журнал команд можно посмотреть, выполнив команду history. Последующее выполнение, например, команды !30 приведёт к выполнению команды, записанной в журнале под номером 30.
Иногда применяют символ ; для разделения строки на последовательно выполняемые команды.
Например, pwd; ls сначала сообщит
нам полное имя текущей директории, а затем покажет её содержимое.
Примеры использования некоторых команд и спецсимволов приведены далее (поиск файлов, содержащих в имени bash_history или пробел).
Команда wc
Команда wc по умолчанию подсчитывает число строк, слов и байтов в указанном файле (FILE). Если указано более одного значения для файла, то результатом подсчёта является общее количество строк. Если файл не указан или указано значение -, то команда считывает стандартный ввод. Словом команда считает последовательность ненулевой длины, состоящую из символов, отделённых пробелами от других слов.
Данная команда имеет несколько полезных опций:
- -c, --bytes - подсчитывает количество байтов;
- -m, --chars - подсчитывает количество символов;
- -l, --lines - подсчитывает количество строк;
- -L, --max-line-length - подсчитывает длину самой длинной строки;
- -w, --words - подсчитывает количество слов;
Например, выполнив команду
wc cdd_bacsu.info
мы получим следующий результат:
2 22 273 cdd_bacsu.info
Т.е. в файле cdd_bacsu.info записано 2 строки, 22 символа и 273 байта.
Файлы wc_help.txt и wc_man.txt содержат информацию о работе с функцией wc.
Найти файл, имя которого содержит "bash_history"
Для нахождения файла следует использовать команду find. Она имеет множество различных опций, из которых нам понадобится лишь -name, так как поиск мы будем усуществлять по имени файла (точнее, по части имени). Используя символ * для маски поиска, составим команду:
find ~ -name '*bash_history*'
После её выполнения мы видим место расположения искомого файла:
/home/students/y11/sapsan/.bash_history
Что же это за файл? Оказывается, bash записывает в историю набранные за сеанс команды, и делает он это только при своём закрытии. Стоит также упомянуть, что по умолчанию bash не дописывает файл .bash_history, а переписывает его. Кстати, историю команд можно посмотреть в терминале простой командой history.
Команда ls по умолчанию игнорирует файлы, начинающиеся с точки. Для того, чтобы просмотреть список всех файлов с помощью команды ls, необходимо использовать параметр -a (или --all). Перейдём в директорию, в которой, как мы вяснили, находится нужный файл. После выполнения команды
ls -a
мы видим список файлов директории, в котором присутствует файл .bash_history.
Найти и переименовать файл с пробелом в имени
Воспользовавшись командой find, осуществим поиск по маске файла:
find ~ -name '* *'
В результате мы получим список имён файлов (с полными путями к ним), содержащих пробел. Выберем, например, файл /home/students/y11/sapsan/video/Sample Pictures.lnk. Скопируем его в папку с текущей работой при помощи команды cp и переименуем так, чтобы пробела в имени не было:
mv Sample\ Pictures.lnk Sample_Pictures.lnk
При помощи команды ls можно убедиться, что файл успешно переименован и теперь не содержит пробела.
∧ НаверхРаздел 2: EMBOSS

infoseq entret showdb seqret matcher stretcher needle water
edialign consambig distmat plotcon
Описание команды infoseq и ее параметров
Информация о команде записана в файл infoseq_help.txt. Как мы видим, эта команда из пакета EMBOSS имеет множество полезных параметров. Рассмотрим некоторые из них.
| Параметр | Применение | Пример | Результат |
| -outfile | Указание имени файла для вывода информации. | infoseq sw:cdd_bacsu -outfile filename | Записывает информацию в файл filename. |
| -columns | С помощью boolean (Y и N) можно применить или отключить оформление вывода в виде колонок. При отключении можно воспользоваться разделением результатов по параметру, указанному с помощью ещё одной опции (см. далее -delimiter). | infoseq sw:cdd_bacsu -columns n | Вывод информации без разделения на колонки, её части отделены друг от друга разделителем | (по умолчанию). См. файл. |
| -delimiter | Выбор разделителя для информации в полученном тексте. Можно использовать любые символы для разделения записи разной информации. | Например, используем пробел в качестве разделителя: infoseq sw:cdd_bacsu -columns n -delimiter ' ' |
Вывод информации без разделения на колонки, её части отделены друг от друга пробелом. См. файл. |
| -html | Форматирование получаемой таблицы как HTML-таблицы. | infoseq sw:cdd_bacsu -html | HTML-таблица с информацией о белке CDD_BACSU из банка SwissProt. См. файл. |
| -heading | Отображение заголовков колонок. | infoseq sw:cdd_bacsu -heading n | Таблица из одной строки с информацией по умолчанию. См. файл. |
Можно "отфильтровать" необходимые нам данные. Для этого существует два способа. Первый - указать, какая информация нам не нужна (-noname, -nodesc и другие; смысл каждой из опций интуитивно понятен, так как все они содержат в своём написании названия колонок получаемой таблицы с информацией об объекте). Второй - указать только необходимую информацию (-only -length, например). Так, выполнив команду
infoseq sw:cdd_bacsu -noname -nohead -notype -nopgc -nodesc -noacc -nousa -nodat -nolen
мы получим только "имя" организма:
Bacillus subtilis
Аналогичный результат можно получить, выполнив более короткую по количеству символов команду:
infoseq sw:cdd_bacsu -only -organism
Результат:
Organism
Bacillus subtilis
Аналогичным образом можно получить колонки name (имя), usa (Uniform Sequence Address последовательности), database (база данных), accession (идентификатор Uniprot AC), type (тип), lenght (число аминокислотных остатков), pgc (процент содержания гуанин-цитозин), organism (название организма), description (описание), а также GI и version (2 вида идентификационных номеров последовательности; по умолчанию эти колонки не отображаются; включить их в выдачу терминала просто: достаточно записать соответственно -gi y и -version y).
Параметры программы infoseq можно разделить на 4 раздела:
- обязательные (это -sequence, например, sw:cdd_bac*);
- дополнительные (это -outfile и -html);
- продвинутые (все остальные параметры, рассмотренные выше, за исключением общих);
- общие (например, -help).
Команда entret
Программа entret на сервере kodomo умеет извлекать записи из локальных банков данных. Чтобы получить документ для белка, необходимо задать в качестве аргумента строку: sw:<ID_белка>. Например, выполнив команду
entret sw:cdd_bacsu cdd_bacsu
мы получим искомую информацию для белка CDD_BACSU, записанную в файле cdd_bacsu.entret.
Рассматриваемая команда имеет два обязательных параметра (указание последовательности [-sequence] и выходного файла [-outfile]), а также продвинутый параметр -firstonly (boolean), необходимый для того, чтобы прочитать только одну последовательность (и затем остановиться).
Команда showdb
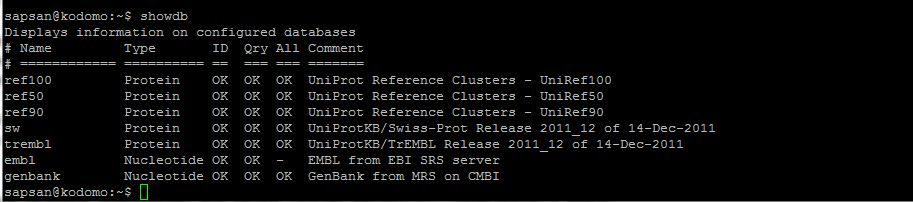
Данная команда отображает информацию о доступных базах данных. Файл showdb_help.txt содержит информацию по использованию данной команды.
Команда showdb не трубует обязательных аргументов. Из дополнительных и продвинутых следует выделить возможность вывода результата как HTML-таблицы (-html) и возможность работы с информацией, которую необходимо включить в выдачу (например, с помощью -full можно отобразить все колонки с информацией о базах данных, а комбинируя параметр -only с такими параметрами, как -heading, -type, -id, -query, -all, -comment, можно добиться отображения только необходимых колонок).
Команда seqret
Команда seqret читает и возвращает запрошенные последовательности (записывает их в файл). Она имеет два обязательных параметра: имя последовательности или её адрес (USA) [-sequence] и имя выходного файла [-outseq]. Продвинутый параметр -firstonly (boolean), позволяет прочитать только одну последовательность (и затем остановиться).
Например, результатом выполнения команды
seqret sw:cdd_bacsu cdd_bacsu.seqret
или
seqret sw:p19079 cdd_bacsu.seqret
будет файл cdd_bacsu.seqret, содержащий запрошенную последовательность в формате .fasta.
Команда matcher
C помощью программы matcher пакета EMBOSS можно получить несколько частичных выравниваний с наибольшим весом.
Рассматриваемая команда имеет три обязательных параметра: два первых - это имя последовательности или её адрес (USA) [-asequence] и [-bsequence],
третий - это имя выходного файла [-outfile].
Есть 4 дополнительных параметра: матрица весов -datafile (по умолчанию -
EBLOSUM62 для белков и EDNAFULL для ДНК), -alternatives для указания количества альтернативных выравниваний (по умолчанию
строится одно выравнивание),
штрафы за открытие и продление гэпа -gapopen и -gapextend (по умолчанию 14 и 4 для белков и 16 и 4 для ДНК соответственно).
Например, в результате выполнения следующей команды
matcher sw:p19079 sw:p32320 cdd_bacsu-cdd_human.matcher -alternatives 5 -gapopen 11 -gapextend 1
мы получим файл cdd_bacsu-cdd_human.matcher, содержащий 5 различных локальных выравниваний с наибольшим весом указанных последовательностей с учётом указанных штрафов за открытие и продление гэпа.
Команда stretcher
C помощью программы stretcher пакета EMBOSS можно получить быстрое оптимальное глобальное выравнивание двух последовательностей по алгоритму Нидлмана — Вунша.
Рассматриваемая команда имеет три обязательных параметра: два первых - это имя последовательности или её адрес (USA) [-asequence] и [-bsequence],
третий - это имя выходного файла [-outfile].
Есть 3 дополнительных параметра: матрица весов -datafile (по умолчанию -
EBLOSUM62 для белков и EDNAFULL для ДНК) и
штрафы за открытие и продление гэпа -gapopen и -gapextend (по умолчанию 12 и 2 для белков и 16 и 4 для ДНК соответственно).
Например, в результате выполнения следующей команды
stretcher sw:p19079 sw:p32320 cdd_bacsu-cdd_human.stretcher -gapopen 11 -gapextend 1
мы получим файл cdd_bacsu-cdd_human.stretcher, содержащий глобальное выравнивание указанных последовательностей с учётом указанных штрафов за открытие и продление гэпа.
Команда needle
Программа needle пакета EMBOSS выдаёт оптимальное полное выравнивание.
Рассматриваемая команда имеет пять обязательных параметров:
два первых - это имя последовательности или её адрес (USA) [-asequence] и [-bsequence];
следующие два - штрафы за открытие и продление гэпа -gapopen и -gapextend (по умолчанию 10.0 и 0.5; можно применить параметр
-auto для использования стандартных значений штрафов);
пятый - это имя выходного файла [-outfile].
Есть 4 дополнительных параметра:
матрица весов -datafile (по умолчанию - EBLOSUM62 для белков и EDNAFULL для ДНК);
-endweight (boolean) для применения штрафов за конечные гэпы;
штрафы за открытие и продление конечных гэпов -endopen и -endextend(по умолчанию 10.0 и 0.5).
Например, в результате выполнения следующей команды
needle sw:p19079 sw:p32320 cdd_bacsu-cdd_human.needle -gapopen 11 -gapextend 1
мы получим файл cdd_bacsu-cdd_human.needle, содержащий оптимальное полное выравнивание последовательностей белков CDD_BACSU (AC P19079) и CDD_HUMAN (AC P32320) с учётом указанных штрафов за открытие и продление гэпа.
Команда water
Программа water пакета EMBOSS выдаёт оптимальное частичное выравнивание.
Рассматриваемая команда имеет пять обязательных параметров:
два первых - это имя последовательности или её адрес (USA) [-asequence] и [-bsequence];
следующие два - штрафы за открытие и продление гэпа -gapopen и -gapextend (по умолчанию 10.0 и 0.5; можно применить параметр
-auto для использования стандартных значений штрафов);
пятый - это имя выходного файла [-outfile].
Есть 1 дополнительный параметр:
матрица весов -datafile (по умолчанию - EBLOSUM62 для белков и EDNAFULL для ДНК).
Например, в результате выполнения следующей команды
water sw:p19079 sw:p32320 cdd_bacsu-cdd_human.water -gapopen 11 -gapextend 1
мы получим файл cdd_bacsu-cdd_human.water, содержащий оптимальное частичное выравнивание последовательностей белков CDD_BACSU (AC P19079) и CDD_HUMAN (AC P32320) с учётом указанных штрафов за открытие и продление гэпа.
Программа edialign
Программа edialign используется для построения множественных выравниваний. Она имеет три обязательных параметра:
- имя набора последовательностей или адрес (USA) [-sequences];
- выходного файла для чтения [-outfile];
- выходного файла с выровненными последовательностями [-outseq].
Среди дополнительных параметров следует обратить внимание на -maxfrag1, указывающий максимальную длину фрагмента последовательности, а также -revcomp, которая позволяет учесть обратную последовательность (и принцип комплементарности).
Из продвинутых парметров интересны -mask, указывающая на замену невыровненных "букв" на символ * вместо их написания в нижнем регистре, и -starnum, позволяющая изменить отображение уровня консервативности с цифр 0 - 9 на *.
Например, в результате выполнения следующей команды
edialign myproteins.fasta myproteins.edialign edialign_alignment.fasta
мы получим файл edialign_alignment.fasta,содержащий выравнивание последовательностей, записанных в файле myproteins.fasta, и файл myproteins.edialign, в котором записана информация о выравнивани.
Программа consambig
Программа consambig используется для обработки множественных выравниваний. Эта программа позволяет получить неоднозначную консенсусную последовательность (т.е. обобщённую последовательность) на основе множественного выравнивания. Она имеет два обязательных параметра: имя файла, содержащего выравнивание, и имя выходного файла.
Среди множества дополнительных параметров, разбитых на группы по отношению к первому или второму обязательному параметрам, могут оказаться наиболее полезными следующие:
- -sbegin1 и -send1, указывающие на начало и конец используемого фрагмента последовательности;
- -sreverse1, указывающая на обратную последовательность (в случае работы с ДНК);
- -snucleotide1 и -sprotein1, указывающие на то, что последовательность соответственно нуклеотидныя или белковая;
- -ossingle2, позволяющая сохранить каждую запись в отдельном файле.
Программа consambig имеет множество других параметров, использование которых рассмотрено в подробном описании программы, получить которое можно, выполнив следующую команду:
tfm consambig
В качестве примера рассмотрим результат выполнения следующей команды:
consambig mafft_alignment.fasta consambig_oufile.fasta
Мы получим файл consambig_oufile.fasta,содержащий консенсусную последовательность, построенную на основе множественного выравнивания, записанного в файле mafft_alignment.fasta.
Программа distmat
Программа distmat используется для создания матрицы эволюционных расстояний для множественного выравнивания. Она вычисляет расстояние между каждой парой последовательностей в множественном выравнивании, при этом возможен выбор различных методов оценки эволюционного расстояния. Выходной файл представляет собой матрицу расстояний для набора последовательностей.
Программа distmat имеет очень подробное описание, котором рассмотрены различные особенности работы с это программой. Получить описание можно, если выполнить следующую команду:
tfm distmat
Обязательные параметры включают в себя имя файла, содержащего выравнивание последовательностей, и имя выходного файла, содержащего матрицу расстояний. Можно также указать метод корректировки множественных замен (-protmethod и -nucmethod).
Среди множества дополнительных параметров, разбитых на группы по отношению к первому или второму обязательному параметрам, могут оказаться наиболее полезными следующие:
- -sbegin1 и -send1, указывающие на начало и конец используемого фрагмента последовательности;
- -sreverse1, указывающая на обратную последовательность (в случае работы с ДНК);
- -snucleotide1 и -sprotein1, указывающие на то, что последовательность соответственно нуклеотидныя или белковая.
Как мы видим, многие параметры сходны с параметрами рассмотренной ранее программы consambig.
В качестве примера рассмотрим результат выполнения следующей команды:
distmat mafft_alignment.fasta myproteins.distmat -protmethod 0
Мы получим файл myproteins.distmat,содержащий матрицу эволюционных расстояний, построенную на основе множественного выравнивания, записанного в файле mafft_alignment.fasta.
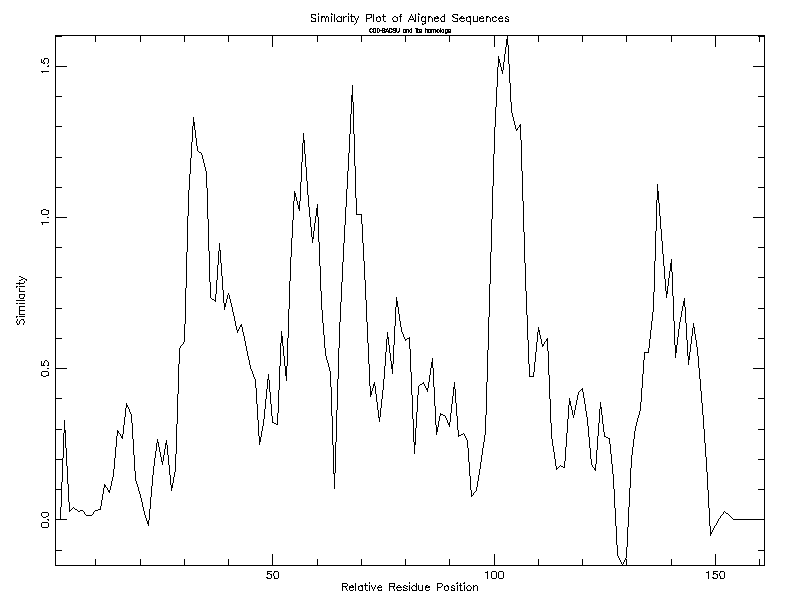
Программа plotcon
Программа plotcon - программа с графической выдачей. Она выполняет построение консервативности выравнивания последовательностей. Программа вычисляет схожесть остатков в определённом участке выравнивания (по всем последовательностям) с учётом весов замен, которые взяты из матрицы схожести.
Программа plotcon имеет очень подробное описание, котором рассмотрены различные особенности работы с это программой. Получить описание можно, если выполнить следующую команду:
tfm plotcon
Обязательные параметры включают в себя имя файла, содержащего выравнивание последовательностей, а также число колонок для вычисления среднего значения степени консервативности (-winsize) и тип графика, который буден получен на выходе (-graph, например, x11 для выдачи картинки на экран монитора, data для получения текстового файла, ps, png, pdf, gif, svg).
Среди множества дополнительных параметров, разбитых на группы по отношению к первому или второму обязательному параметрам, могут оказаться наиболее полезными следующие:
- -sbegin1 и -send1, указывающие на начало и конец используемого фрагмента последовательности;
- -sreverse1, указывающая на обратную последовательность (в случае работы с ДНК);
- -snucleotide1 и -sprotein1, указывающие на то, что последовательность соответственно нуклеотидныя или белковая;
- -gtitle для указания заголовка графика;
- -gsubtitle для указания подзаголовка графика;
- -gxtitle и -gytitle для указания названий осей.
Как мы видим, многие параметры (за исключением тех, которые непосредственно относятся к выходному файлу) сходны с параметрами рассмотренных ранее программ consambig и distmat.
В качестве примера рассмотрим результат выполнения следующей команды:
plotcon mafft_alignment.fasta -graph png -winsize 4 -gsubtitle "CDD_BACSU and its homologs"
Мы получим файл plotcon.1.png, представляющий собой график консервативности, построенный на основе множественного выравнивания, записанного в файле mafft_alignment.fasta:
Получение информации о своём белке
Команда infoseq sw:protein_id выдает на терминал информацию о записи protein_id банка SwissProt.
Идентификатор моего белка - CDD_BACSU.
С помощью команды infoseq sw:cdd_bacsu можно получить информацию о моём белке.
Записать информацию в файл можно, перенаправив stdout в файл:
infoseq sw:cdd_bacsu > cdd_bacsu.info
Таким образом, в файле cdd_bacsu.info записана полученная информация о белке.
Запись информации о программе infoseq в файл
Команда infoseq -help выдает на терминал информацию о программе infoseq. Несложно записать эту информацию (поток stderr) в файл:
infoseq -help 2> infoseq_help.txt
Теперь файл infoseq_help.txt содержит информацию о программе infoseq.
Нахождение белков CDD в других видах рода Bacillus.
Для выполнения задания необходимо использовать символ *. Запишем команду:
infoseq sw:cdd_bac*
Она выдает на терминал искомую информацию:
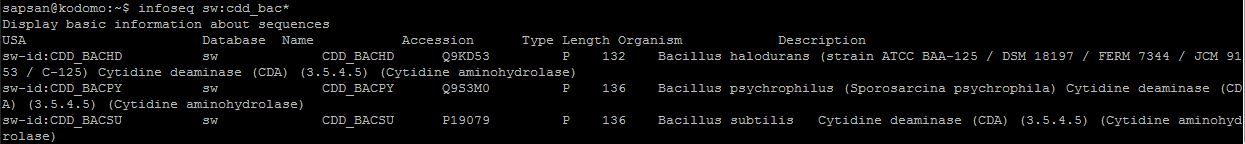
Мы видим, что белок с кодом CDD содержится в трех видах рода Bacillus. Запишем полученные данные в файл:
infoseq sw:cdd_bac* > cdd_in_bacillus_genus.txt
Теперь в файле cdd_in_bacillus_genus.txt записана искомая информация.
Безусловно, можно использовать различные опции изменения результата выдачи. Например, если нам необходимо получить только названия организмов рода Bacillus, в которых встречается данный белок, можно записать:
infoseq sw:cdd_bac* -only -organism
Результат будет выглядеть следующим образом:
Organism
Bacillus halodurans (strain ATCC BAA-125 / DSM 18197 / FERM 7344 / JCM 9153 / C-125)
Bacillus psychrophilus (Sporosarcina psychrophila)
Bacillus subtilis
Ещё один пример: выполнив команду
infoseq sw:cdd_bac* -nousa -nodat -notype -heading n -outfile cdd_in_bacillus_genus_example.txt
получим следующий файл с соответствующим выполненной команде содержимым.
Подробнее о различных опциях программы написано выше.
Ссылки
- Информация об использовании команды infoseq.
- Информация о белке cdd_bacsu.
- Информация о белках cdd в видах рода Bacillus.