|
|
Я работала с чтениями последовательности пятой хромосомы человека и с пятой хромосомой из сборки h19.
Сначала с помощью программы FastQC
был проведен контроль качества данных чтений. Программу можно запустить командой fastqc chr5.fastq. Результат работы программы -
набор картинок, иллюстрирующих качество чтений на основании множества параметров. Все изображения можно посмотреть по ссылке
chr5_fastqc.
На рисунке 1 представлены основные характеристики файла с чтениями, такие как число последовательностей, разброс их длины и средний GC-состав.
Наиболее показательно качество чтений иллюстрирует распределение качества по нуклеотидам (per base sequence quality). Это изображение представлено
на рисунке 2.
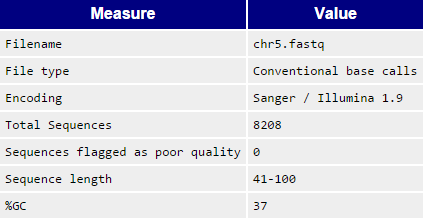
Рисунок 1. Общая информация о наборе чтений.
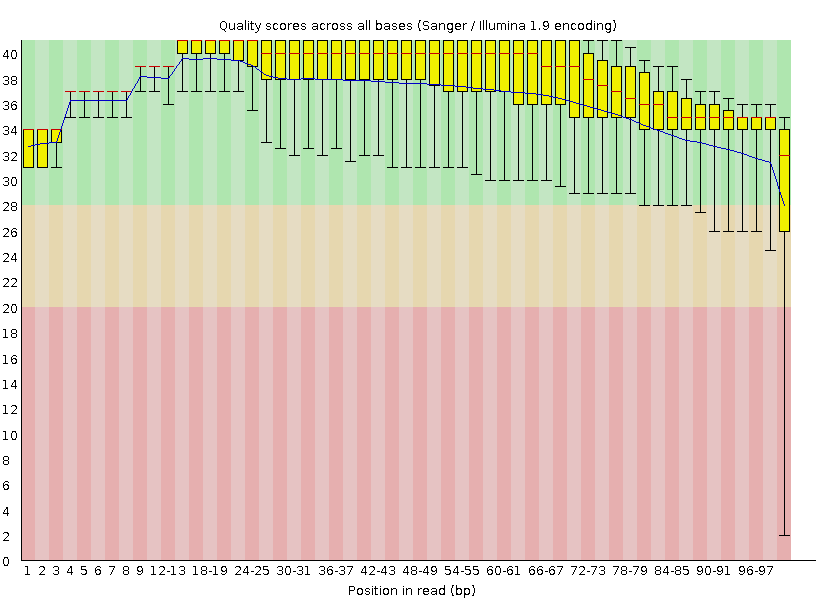
Рисунок 2. Качество нуклеотидов в чтениях ("Per base sequence quality"). По горизонтальной оси отложена позиция в последовательности
(в п.о.), по вертикальной оси - разброс качества по всем чтениям.
Далее я произвела очистку чтений, отрезав с конца нуклеотиды с качеством ниже 20 и оставив только последовательности, длина которых составляет более
50 нуклеотидов. Для этого была использована программа Trimmomatic.
Для проведения такой очистки была использована команда
java -jar /usr/share/java/trimmomatic.jar SE -phred33 infile.fastq outfile.fastq TRAILING:20 MINLEN:50. Результат работы программы представлен
на рисунке 3.
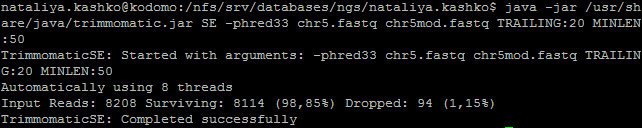
Рисунок 3. Результат работы программы Trimmomatic.
Как видно из рисунка 3, Trimmomatic отсеял 94 последовательностей (1.15%). Из них 2 было удалено после проведения TRAILING (отрезание нуклеотидов с
плохим качеством с конца), а остальные - из-за несоответствующей длины (меньше 50 п.о.). По рисунку 1 видно, что в исходном файле присутствовали чтения
с длиной от 41 п.о. В обработанном файле такие последоватеьности были удалены.
Качество очищенных чтений также было проанализировано программой FastQC. Все полученные иллюстрации доступны по ссылке
chr5mod_fastqc. На рисунках 4 и 5 приведены изображения, аналогичные рисункам 1 и 2, только для очищенных чтений.
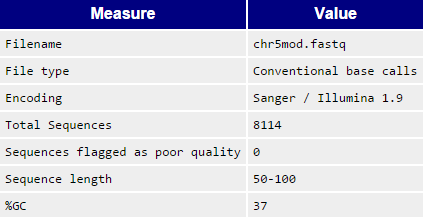
Рисунок 4. Общая информация о наборе чтений после очистки с помощью программы Trimmomatic.
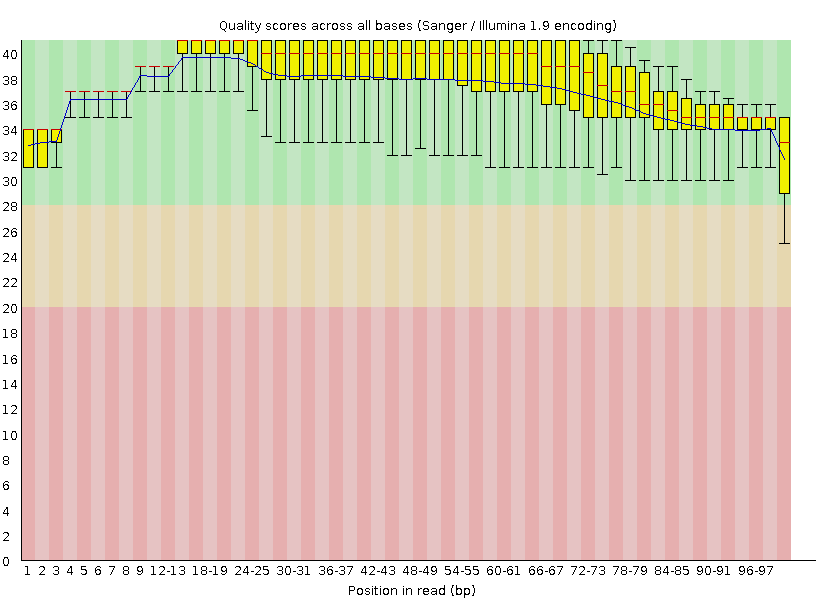
Рисунок 5. Качество нуклеотидов в чтениях после очистки.
Из рисунков 1 и 4 видно, что исходное число чтений составляло 8208, а после чистки чтений стало 8114. Подтверждение того, что среди очищенных чтений остались только
те, длина которых более 50 п.о., представлено на картинках "Sequence Length Distribution". Результат отрезания нуклеотидов с плохим качеством с конца последовательности
представлен на рисунках 2 и 5. Если посмотреть на последний столбик, то видно, что среднее качество при очистке изменилось с 28 до 32, а нижняя граница - с 2 до 25.
Затем я приступила к картированию чтений. В качестве референсной последовательности я использовала последовательность пятой хромосомы человека из сборки h19.
Сначала я проиндексировала референсную последовательность с помощью программы BWA. Для этого
использовалась команда bwa index chr5.fasta. Затем было построено вырванивание очищенных чтений с проиндексированной референсной последовательносью командой
bwa mem chr5.fasta chr5mod.fastq > chr5.sam.
С помощью пакета samtools полученный файл chr5.bam был переведен в файл формата .sam. Для этого была использована команда
samtools view chr5.sam -b -o chr5.bam. Опция -b задает формат выходного файла .bam, опция -o позволяет указать его название.
Полученный .bam файл с выравниваниями чтений с референсом был отсортирован по координате начала чтения в референсе с помощью команды
samtools sort -T /tmp/chr5_sorted -o chr5_sorted.bam chr5.bam. Затем отсортированный файл был проиндексирован командой samtools index chr5_sorted.bam.
С помощью команды samtools idxstats chr5_sorted.bam было посчитано количество чтений, откартированных на хромосому. На рисунке 6 представлены результаты работы
этой программы - табличка, показывающая, что на референсную пятую хромосому откартировалось 8113 чтений (все, кроме одного). Также в таблице указана длина пятой хромосомы,
равная 180,915,260 п.о. Что означает цифра 2 в нижней строке я, к сожалению, не смогла понять.
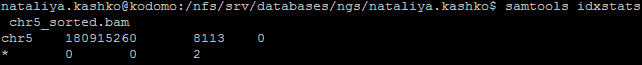
Рисунок 6. Результат работы программы samtools idxstats.
С помощью команды samtools mpileup -uf chr5.fasta chr5_sorted.bam -o chr5snp.bcf был создан файл с полиморфизмами в формате .bcf. Затем этот файл был переведен в файл в формате
.vcf командой bcftools call -cv chr5snp.bcf -o chr5snp.vcf. Файл chr5snp.vcf содержит список отличий между референсом и чтениями.
Всего найдено 32 полиморфизма, среди которых 4 инделя и 28 замен одного нуклеотида. В таблице 1 представлена информация о некоторых из них. Также эти замены были визуализированы с
помощью программы IGV. Изображения представлены на рисунках 6-9.
|
Таблица 1. Информация о четырех полиморфизмах, найденных при выравнивании чтений с референсной последовательностью - пятой хромосомой человека.
|
| Координата |
Тип полиморфизма |
Референс |
Чтения |
Глубина покрытия |
Качество чтений |
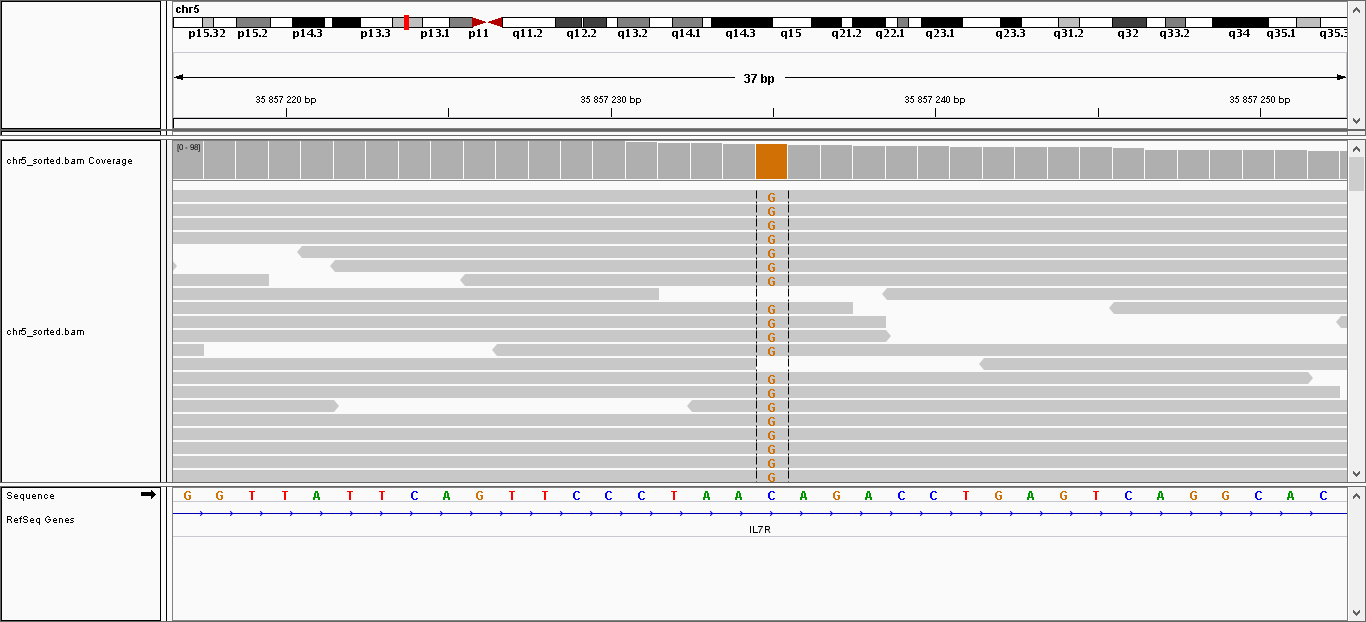
| 35,857,235 |
Замена |
C |
G |
86 |
221.999 |
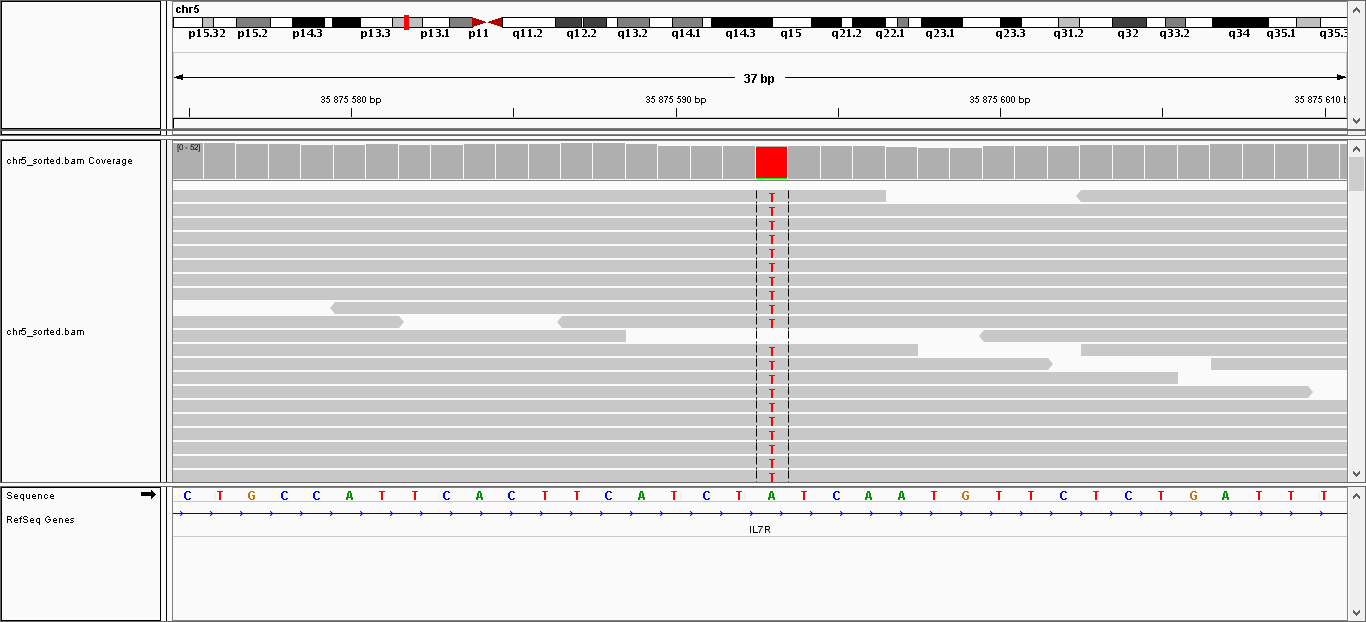
| 35,875,593 |
Замена |
A |
T |
43 |
221.999 |
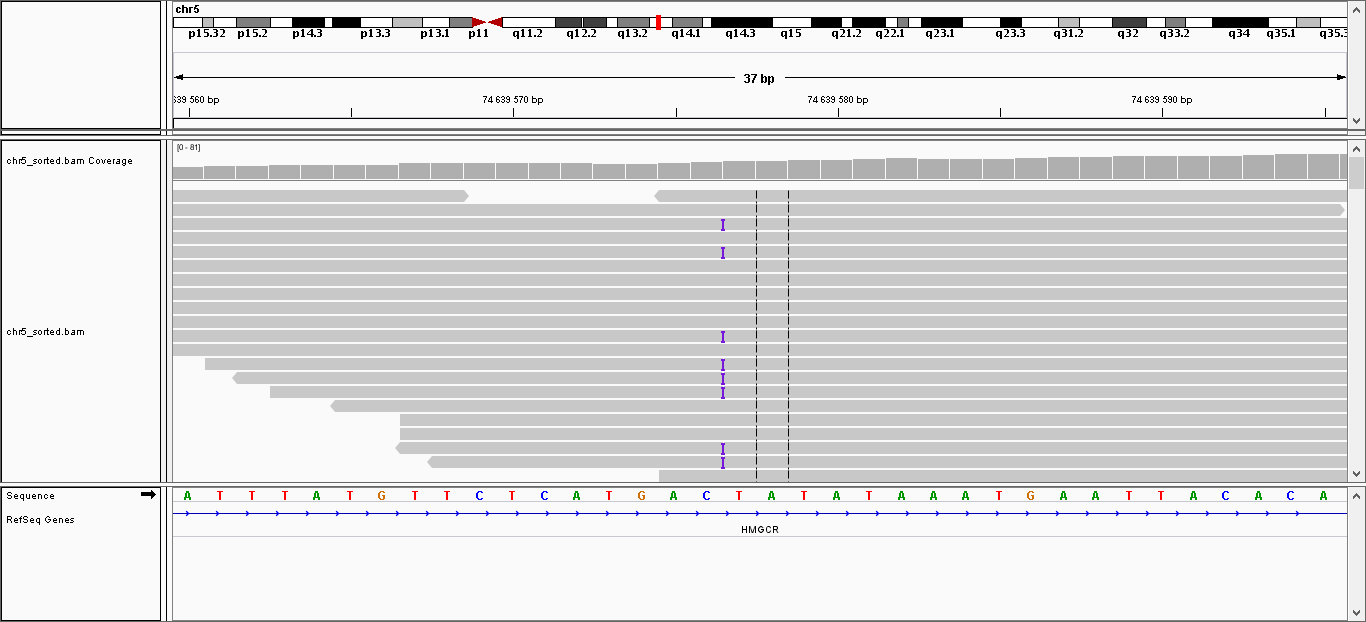
| 74,639,576 |
Вставка |
C |
CAA |
36 |
217.468* |
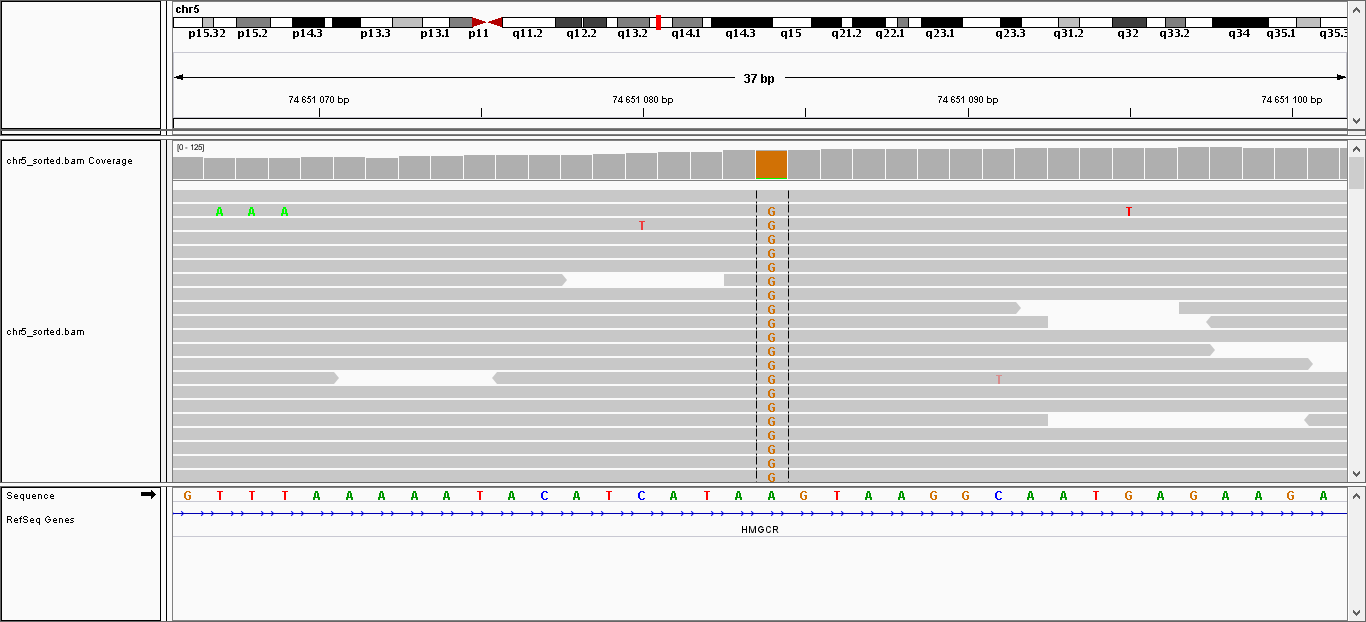
| 74,651,084 |
Замена |
A |
G |
91 |
221.999 |
* - указано, что число чтений, подтверждающих данную замену, равно 12
Рисунок 6. Изображение замены C на G в интроне гена IL7R. Столбик выделяет замены в чтениях, снизу указана референсная последовательность. Верхняя панель
отображает положение на пятой хромосоме. Изображение получено с помощью программы IGV.
Рисунок 7. Изображение замены A на T в интроне гена IL7R.
Рисунок 8. Изображение вставки (буква I, вставка AA) в интрон гена HMGCR. Вставка произошла между нуклеотидами C и T в референсе.
Рисунок 9. Изображение замены A на G в интроне гена HMGCR.
Полученный список SNP (без инделей) я проаннотировала с помощью программы Annovar
с использованием пяти баз данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar. Для этого я использовала команды, представленные в таблице 2.
По результатам анализа с помощью базы RefGene было обнаружено 16 гомозиготных замен и 12 гетерозиготных (hom и het соответственно). SNP при аннотации по базе данных RefGene классифицируются по положению, которое они
занимают в геноме, то есть попадают ли они в экзоны или интроны генов, в межгенные области, в гены некодирующих РНК или в 3'- и 5'-некодирующие области (UTR3 и UTR5). Из описываемых SNP 4 попадают в экзоны,
1 - в UTR3 и остальные 23 - в интроны.
Замены попали в гены IL7R, CAPSL и HMGCR, однако затронули экзоны только первых двух генов из перечисленных. Все замены в экзонах несинонимичные, то есть они приводят к замене аминокислоты
в белковой последовательности.
Ген CAPSL кодирует кальций-связывающий белок кальцифозин. Замена C на T приводит к замене аргинина на глутамин в белке (R85Q), однако по данным UniProt такая замена является естественным
полиморфизмом (ссылка: CAPSL_HUMAN).
Ген IL7R кодирует альфа-цепь рецептора интерлейкина-7. Показано, что этот белок участвует в рекомбинации вариаебльных участков в генах иммуноглобулинов при развитии лимфоцитов. Проведенные исследования
в мышах показывают, что этот белок необходим для блокировки апоптоза при дифференциации Т-лимфоцитов. Мутации в этом белке могут приводить к развитию тяжелого комбинированного иммунодефицита (SCID), а также
некоторых заболеваний, таких как Т-клеточная острая лимфобластная лейкимия, рассеянный склероз, ревматоидный артрит и ювенильный идиопатический артрит.
Полный список нуклеотидных замен представлен в таблице excel: snp.xlsx. Замены в экзонах привели к следующим аминокислотным заменам: в гене IL7R I66T, V138I, T244I; в гене CAPSL R85Q.
Такой формат записи означает, что аминокислота слева (первая буква), находящаяся в позиции, номер которой указан в центре, заменена на аминокислоту справа.
Из найденных SNP rs имеют 24 штуки, не имеют - 4. rs - это идентификатор записи данной замены в базе данных dbSNP. Таким образом, в этой базе не аннотировано всего 4 замены. В основном не имеют rs SNP с очень низким
качеством и покрытием. В целом, замен с качеством меньше 20 найдено 4, с покрытием меньше 20 - 11.
Клиническое описание SNP предоставляют базы данных Clinvar и GWAS. Clinvar выделяет 3 замены в гене IL7R, указанные выше, из которых первые 2 являются патогенными и приводят к SCID, а третья является неохарактеризованной.
База GWAS описывает 5 замен, из которых одна также аннотирована в Clinvar и была выявлена при исследовании рассеянного склероза и диабета первого типа. Вторая замена - уже упоминавшийся естественный полиморфизм, который также,
по данным GWAS, имеет отношение к диабету первого типа. Остальные три SNP относятся к интронным участкам гена HMGCR и к его 3'-некодирующей области и были обнаружены при изучении уровней холестерина.
Результаты по всем базам данных представлены в сводной таблице excel (snp.xlsx). Все использованные в практикуме команды описаны в таблице 2.
|
Таблица 2. Команды, использованные в практикуме, для описания полиморфизмов человека.
|
| Команда |
Что делает |
| fastqc chr5.fastq |
анализирует качество чтений |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 infile.fastq outfile.fastq TRAILING:20 MINLEN:50 |
очищает чтения: сначала отрезает с конца каждого чтения нуклеотиды с качеством ниже 20, затем удаляет чтения длиной меньше 50 нуклеотидов |
| bwa index chr5.fasta |
индексирует референсную последовательность (пятую хромосому человека) |
| bwa mem chr5.fasta chr5mod.fastq > chr5.sam |
строит выравнивание чтений с референсной последовательностью, то есть картирует все чтения по хромосоме |
| samtools view chr5.sam -b -o chr5.bam |
переводит выравнивание чтений с референсом в бинарный формат, с которым потом работают программы |
| samtools sort -T /tmp/chr5_sorted -o chr5_sorted.bam chr5.bam |
сортирует выравнивание чтений с референсом по координате начала чтения в референсе |
| samtools index chr5_sorted.bam |
индексирует отсортированный файл |
| samtools idxstats chr5_sorted.bam |
определяет, сколько чтений откартировалось на хромосому |
| samtools mpileup -uf chr5.fasta chr5_sorted.bam -o chr5snp.bcf |
создает файл с полиморфизмами |
| bcftools call -cv chr5snp.bcf -o chr5snp.vcf |
переводит полученный .bcf файл в список отличий между референсом и чтениями в формате .vcf. |
| perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 chr5snp.vcf -outfile chr5snp.avinput * |
создает на основе файла .vcf файл, с которым может работать annovar |
| perl */annotate_variation.pl -geneanno -dbtype refGene -buildver hg19 chr5snp.avinput -outfile chr5snp_refgene */humandb/ |
аннотирует файл со списком SNP по базе данных refgene с использованием сборки генома человека h19 |
| perl */annotate_variation.pl -filter -dbtype snp138 -buildver hg19 chr5snp.avinput -outfile chr5snp_snp138 */humandb/ |
аннотирует файл со списком SNP по базе данных dbsnp |
| perl */annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 chr5snp.avinput -outfile chr5snp_1000g2014oct */humandb/ |
аннотирует файл со списком SNP по базе данных 1000genomes |
| perl */annotate_variation.pl -regionanno -dbtype gwasCatalog -buildver hg19 chr5snp.avinput -outfile chr5snp_gwas */humandb/ |
аннотирует файл со списком SNP по базе данных GWAS |
| perl */annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 chr5snp.avinput -outfile chr5snp_clinvar */humandb/ |
аннотирует файл со списком SNP по базе данных Clinvar |
* - далее адрес /nfs/srv/databases/annovar будет обозначаться символом *
|