|
|||||||||||||||||
|
|
С помощью пакета программ velvet я собрала из чтений, использованных в предыдущем практикуме по ресеквенированию, контиги без использования
референсной последовательности. Использованный файл с чтениями (chr5mod.fastq) был очищен программой Trimmomatic. Сначала из файла были выделены k-меры
программой velveth (команда velveth chr5 31 -fastq -short chr5mod.fastq), затем была проведена сборка контигов (команда velvetg chr5). chr5 - это название директории, с которой
работает программа. Полученный набор контигов представлен в файле contigs.fasta.
Далее я построила выравнивание получившихся контигов с хромосомой с помощью программы blastn (алгоритм megablast). Сначала из последовательности хромосомы была создана база для работы blast (команда makeblastdb -in chr5.fasta -dbtype nucl -out chr5), затем был произведен поиск всех контигов в этой базе (команда blastn -query contigs31.fasta -db chr5 -outfmt 6 -out blast.out). Все находки представлены в excel-файле chr5.xlsx (лист k31 alignment). Также там описаны те контиги, которые картируются на хромосому единственным образом (лист k31 contigs). Всего было найдено 168 контигов, из которых 164 - по одному разу. Контиг 108 найден 823 раза, контиг 109 - 11 раз, контиг 258 - 2172 раза и контиг 81 - 479 раз. Возможно, это связано с тем, что данные контиги представляют распространенные в геноме повторяющиеся участки или мобильные элементы генома. При значении k=31 есть множество перекрываний контигов, однако, несмотря на это, эти перекрывающиеся контиги не объединяются в один. В неготорых случаях это можно объяснить тем, что длина перекрывающегося участка меньше длины k-мера (то есть меньше 31), поэтому данный контиг не может быть продолжен по графу для такого значения k. Более длинные перекрывания встречаются между контигами, которые действительно являются продолжением друг друга, но при этом ошибки в секвенировании приводят к небольшим различиям между последовательностями контигов и, следовательно, программа не может их объединить. Пример таких контигов представлен на рисунках 1a и 1b. И, наконец, длинные перекрывания встречаются между контигами, которые выравниваются с разными цепями хромосомы. Эти контиги неправильно было бы объединять, так как они представляют собой комплементарные цепи. 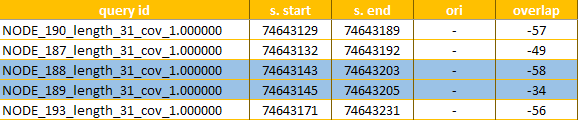
Рисунок 1a. Фрагмент таблицы excel (chr5.xlsx) с выделенными контигами 188 и 189, расположенными на комплементарной цепи и перекрывающимися на 58 п.о. 
Рисунок 1b. Фрагмент fasta-файла с контигами (contigs.fasta) с последовательностями контигов 188 и 189. Выделены различающиеся участки в этих последовательностях (все остальные буквы совпадают). Различия могли возникнуть из-за ошибок при секвенировании или при обработке результатов секвенирования. Разрывы между контигами можно объяснить тем, что данные чтения содержат только некоторые гены, закодированные на пятой хромосоме. Пространства между генами и другие некодирующие области не могут быть покрыты данными контигами, так как эти области просто не секвенировались. Если рассмотреть выравнивание контигов, полученных для значения k=25, с хромосомой (лист k25 contigs), то мы увидим, что все перекрывания контигов, расположенных на одной цепи, имеют длину 23 п.о. и меньше. Это дополнительно подтверждает предположение, что для объединения контигов необходимо перекрывание длины k и больше. |
||||||||||||||||
|
© Наталия Кашко, 2015 |
|||||||||||||||||