|
|
Упражнения
-
Несколько файлов в формате fasta собрать в единый файл
Сначала я создала файл с названиями записей в USA-формате (listfile). Для этого я использовала команду ls > list (названия fasta-файлов из определенной
директории были записаны в файл list). Затем я объеденила эти файлы в один с помощью программы seqret: seqret @list sequences.fasta. В результате применения
этой команды последовательности из файлов, названия которых были записаны в файле list, объединились в файл sequences.fasta.
-
Один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы
Я использовала команду seqretsplit coding2.fasta seq.fasta. В файле coding2.fasta записано несколько последовательностей. Программа seqretsplit
записывает каждую из этих последовательностей в отдельный файл. В результате получается столько файлов (в формате fasta), сколько последовательностей было в исходном файле.
-
Из файла с хромосомой в формате .gb вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле
Последовательность хромосомы имеет AC U00096 в базе данных GenBank, поэтому к ней можно обращаться следующим образом: genbank:U00096. Для того, чтобы вырезать
кодирующие последовательности, я сначала выбрала три CDS из данного файла. Далее я создала listfile с координатами этих CDS (например, с помощью команды echo). В этом файле были
такие записи:
genbank:U00096[5683:6459:r]
genbank:U00096[15445:16557]
genbank:U00096[108279:110984]
Затем я объеденила эти последовательности в один файл, как в упражнении 1. Команда: seqret @list cds.fasta. В результате в файле cds.fasta оказалось 3 последовательности,
каждая из которых начиналась на ATG и заканчивалась на стоп-кодон (в данном случае TAA).
-
Транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода. Результат - в одном fasta файле
Я использовала файл coding2.fasta с кодирующими последовательностями. Так как все эти последовательности начинались со старт-кодона, я использовала опцию -frame 1. Вся команда целиком:
transeq coding2.fasta proteins.fasta -frame 1. Использованная таблица - стандартная таблица генетического кода. В результате в файле proteins.fasta оказалось столько же
последовательностей белков, сколько было нуклеотидных последовательностей в файле coding2.fasta. Стоп-кодон транслируется как *
-
Транслировать данную нуклеотидную последовательность в шести рамках
Чтобы транслировать последовательность во всех шести рамках, надо использовать опцию -frame 6. Команда: transeq coding.fasta frames.fasta -frame 6. Результат - в файле
frames.fasta оказалось 6 белковых последовательностей, полученных при транслировании одной последовательности coding.fasta в 6 рамках.
-
Перевести выравнивание и из fasta формате в формат .msf
Для изменения формата файла используется программа seqret. Новый формат задается с помощью ::, при этом формат указывается до этого символа. Команда выгялдит так:
seqret alignment.fasta msf::alignment.msf. В результате мы получаем файл alignment.msf, в котором указана дополнительная информация о выравниваемых последовательностях и выравнивание
всех последовательностей (а не выровненные последовательности по отдельности, как в формате .fasta).
-
Выдать в выходной поток число совпадающих букв между второй последовательностью выравнивания и всеми остальными (на выходе только имя последовательности и число)
Чтобы получить информацию о выравнивании, используется программа infoalign. Так как нам надо получить только название последовательности и число идентичных букв, нужно использовать
опцию -only: infoalign alignment.fasta align.infoalign -only -name -idcount. Далее нужно взять только вторую строку из полученной таблицы. Это можно сделать с помощью команды sed -n 2p
или head -2 | tail -1 с указанием имени выходного файла align.infoalign в конце. В резульате мы получаем строку таблицы, в которой записано название второй последовательности и число
позиций, совпадающих с позициями во всех остальных последовательностей.
-
Перевести аннотации особенностей в записи формата .gb в табличный формат .gff
Я использовала команду featcopy chromosome.gb table.gff. Полученная таблица может быть отформатирована в Excel. В ней указаны границы генов и CDS (с какого по какой нуклеотид, ориентация цепи),
а также все остальные поля и квалификаторы из раздела feature table.
-
Из данного файла с хромосомой в формате .gb получить fasta файл с кодирующими последовательностями; (*) добавить в описание каждой последовательности функцию белка (из поля product)
Получение отдельных элементов из записи в формате .gb можно осуществить с помощью программы extractfeat. К сожалению, я не смогла выполнить задание для данного файла с хромосомой, поэтому
я использовала файл NC_019684.gbk - аналогичную запись для хромосомы из бактерии Nostoc sp., с которой я в дальнейшем работала в практикуме. Использованная команда для извлечения
кодирующих последовательностей: extractfeat NC_019684.gbk -type CDS cds.fasta. Опция type указывает, какой тип данных из поля feature table следует извлечь. В файле cds.fasta записаны
все кодирующие последовательности из данной записи. Чтобы добавить в описание функцию получающегося белка, следует дополнительно добавить опцию describe (в результате команда выглядит так:
extractfeat NC_019684.gbk -type CDS -describe product cds.fasta).
-
Перемешать буквы в данной нуклеотидной последовательности; (*) проверить с помощью blastn сколько "достоверных" находок (с E-value < 0.1) найдется в нуклеотидном банке данных
С помощью команды shuffle -o shuffled.fasta coding.fasta я перемешала буквы в последовательности coding.fasta и записала полученный результат в файл shuffled.fasta. Поиск в
blastn не выдал ни одной достоверной находки (наименьшее E-value было 2.8), однако в получившейся последовательности оказался участок длиной около 30 нуклеотидов, который очень
похож на участок из генома вируса герпеса человека (процент идентичных позиций - 94%). Из-за такого совпадения было найдено 113 похожих последовательностей, 109 из которых принадлежат
данному вирусу. Однако query cover составляет всего 1%, поэтому судить о достоверности этих находок сложно.
-
Найдите частоты кодонов в данных кодирующих последовательностях
Данное задание можно выполнить с помощью программы cusp. Команда следующая: cusp coding2.fasta frequency.cusp. Файл frequency.cusp представляет собой таблицу, в которой
указаны кодоны, какие аминокислоты они кодируют, сколько было найдено кодонов каждого типа и какой процент от общего числа они составляют (frequency).
-
Найдите частоты динуклеотидов в данной нуклеотидной последовательности и сравните их с ожидаемыми
Использована команда compseq coding.fasta -word 2 -calcfreq double.compseq. Опция -word задает количество букв, показывающее, частоту участков какой длины следует посчитать. -calcfreq
позволяет рассчитать ожидаемые частоты, а также сравнивает их с реальными (используя отношение реальных часток к предсказанным). Результат - файл с расширением .compseq - таблица частот по всем
динуклеотидам. Практически для всех наблюдаемая частота встречаемости несильно отличается от ожидаемой (частное составляет 1±0.2). У двух динуклеотидов (TA и AG) этот показатель примерно равен
0.6-0.65, то есть в реальности они встречаются реже, чем было предсказано. Однако это может быть связано с небольшим количеством данных, например, с особенностями исследуемой последовательности.
-
Выровняйте кодирующие последовательности соответственно выравниванию белков - их продуктов
Команда tranalign gene_sequences.fasta protein_alignment.fasta align.fasta. Результат - выровненные нуклеиновые последоватльности в формате fasta. Программа tranalign использует
имеющееся выравнивание для белков (как для более консервативных биополимеров) для выравнивания кодирующих их генов.
Сравнение аннотации генов белков в хромосоме бактерии Nostoc sp. с трансляциями длинных открытых рамок считывания
Для выполнения данного задания я взяла хромосому бактерии Nostoc sp. (подробнее про нее можно прочитать в практикуме из
первого семестра). Хромосома у этой бактерии одна (AC в GenBank CP003552, в RefSeq NC_019684), также имеются записи с последовательностями двух плазмид.
Сначала я получила файл с трансляциями длинных открытых рамок с помощью программы getorf. Я использовала следующую команду:
getorf NC_019684.gbk orfs.fasta -table 11 -minsize 180 -find 1 -circular. Опция -table задает таблицу генетического кода, в данной случае 11 (бактериальная таблица генетического кода). Опция
-minsize указывает, какова должна быть минимальная длина открытой рамки. Опция -find 1 позволяет транслировать найденные рамки от старт-кодона до стоп-кодона. Опция -circular означает, что данная
молекула ДНК имеет кольцевую структуру.
Для получения списка координат и ориентаций найденных открытых рамок я использовала программу infoseq. Команда:
infoseq orfs.fasta -outfile info -only -accession -length -description. Эта программа выдает информацию о полученном файле orfs.fasta, а именно длину последовательности белка, ее название (в accession
указан номер рамки) и описание (с указанием координат в геноме). Данные представлены в виде таблицы, файл info можно импортировать в Excel. Получившаяся таблица доступна по ссылке:
orfs.xlsx, трансляции открытых рамок отсортированы по их началу в геноме.
Список аннотированных белков был получен из файла NC_019684.ptt. В итоговой таблице имеются следующие столбцы: locus_tag, start, end, ori, length, PID, product. Таблица доступна по
proteins.xlsx, гены белков отсортированы по началу в геноме. Последовательности всех белков можно посмотреть в файле proteins.fasta,
он получен их файла NC_019684.faa.
Далее я произвела сравнение двух таблиц Excel. Сразу можно сказать, что таблицы получились разными хотя бы потому, что открытых рамок было найдено 16771, а белков аннотировано 5355. Эти различия могут быть обусловлены
тем, что не каждая открытая рамка что-то кодирует, например, из-за относительно небольшой заданной минимальной длины рамки (180 п.о.) или из-за их перекрывания, что препятствует нормальному синтезу белков. Большинство
реальных белков было найдено трансляцией открытых рамок, однако точное соответствие начала и конца кодирующих последовательностей встречается редко. Чаще всего
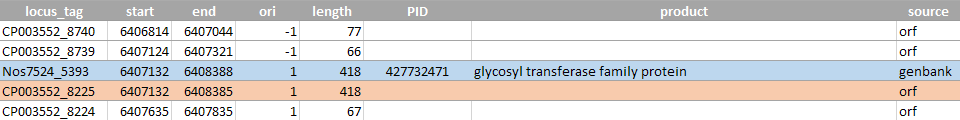
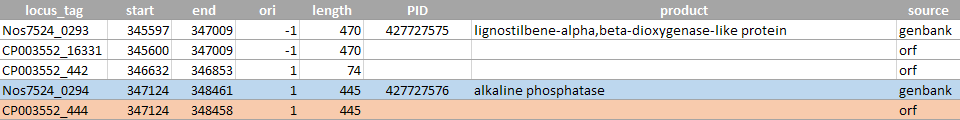
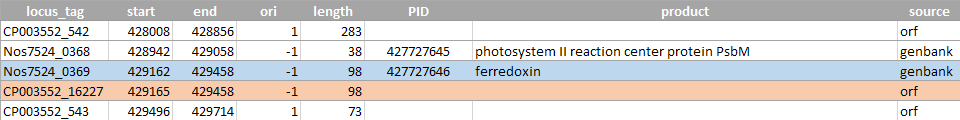
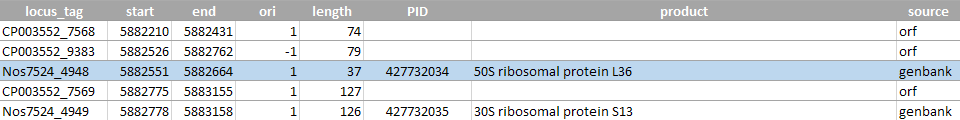
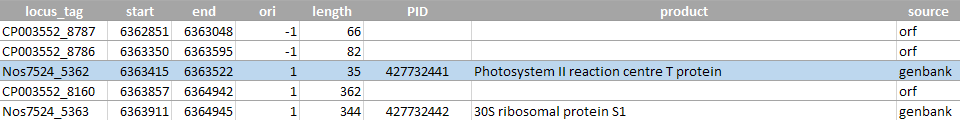
открытая рамка оказывается сдвинута на 3 нуклеотида в одну или в другую сторону (рисунки 1-4). Такое различие может иметь смысл, так как 3 нуклеотида составляют один кодон. Возможно, открытые рамки учитывают стоп-кодоны
(по крайней мере, при поиске рамок с заданными таким образом параметрами), а в аннотированных белках их нет. Некоторые белки (в основном гипотетические) не были найдены с помощью программы getorf из-за того,
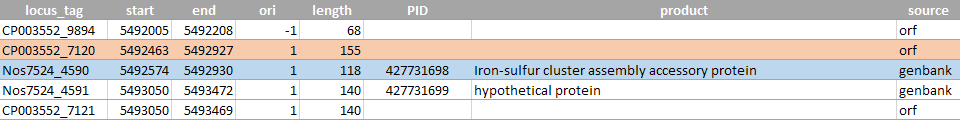
что установленный порог длины соответствует белкам размером более 60 а.о., а реальные белки могут иметь длину около 30 а.о. (рисунки 5, 6). Иногда открытая рамка и ген начинаются в одном месте, но ген заканчивается раньше рамки.
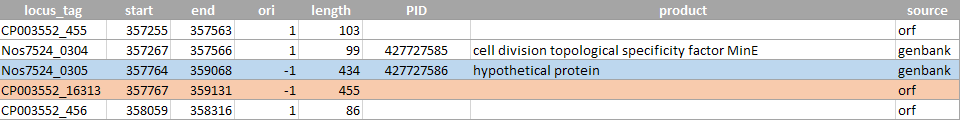
Я выбрала 4 примера таких различий (рисунки 7-10) и проанализировала нуклеотидные последовательности из генома в области этих генов. Расширенная таблица генетического кода для бактерий включает нестандартные старт-кодоны,
такие как GTG, CTG, TTG, ATT (помимо обычного ATG). Найденные рамки оказались более длинными, чем гены, так как, по-видимому, алгоритм поиска рамок выдает самую длинну из возможных рамок в этом месте генома, не учитывая
тот факт, что другие старт-кодоны встречаются реже, чем ATG. В каждом из четырех примеров программа нашла более длинную рамку, начинающуюся с какого-то из нестандартных кодонов, а аннотированный ген начинается с ATG. Также
для одного из таких случаев приведена последовательность в данном участке генома с отмеченными старт-кодонами открытой рамки и гена (рисунок 11). Интересно, что многие рамки начинаются с кодона ATA. Объяснения этому факту я
найти не могу, так как этот кодон не является стартовым в используемой таблице генетического кода.
Рисунок 1 (сверху). Пример открытой рамки и гена, последовательности которых сдвинуты на 3 нуклеотида относительно друг друга. Ген и рамка расположены на прямой цепи. Возможное объяснение: программа
getorf не учитывает стоп-кодоны в конце открытой рамки при указании координат рамки в геноме. Голубым отмечен аннотированный ген, красным - соответствующая ему открытая рамка.
Рисунок 2 (снизу). Еще один пример различия такого типа.
Рисунок 3 (сверху). Пример открытой рамки и гена, последовательности которых сдвинуты на 3 нуклеотида относительно друг друга. Ген и рамка расположены на обратной цепи. В этом случае ситуация аналогична
вышеизложенной: программа getorf не учитывает стоп-кодоны, а в аннотированных генах он, наоборот, учитывается (начало на позиции 2383570, последовательность открытой рамки заканчивается раньше,
чем последовательность гена). Голубым отмечен аннотированный ген, красным - соответствующая ему открытая рамка.
Рисунок 4 (снизу). Еще один пример различия такого типа.
Рисунок 5 (сверху). Пример аннотированного белка, который не был найден программой getorf из-за слишком высокого порога размера открытой рамки (180 п.о., что соответствует белкам от 60 а.о.).
Голубым отмечен аннотированный ген.
Рисунок 6 (снизу). Еще один пример различия такого типа.
Рисунок 7 (самый верхний). Значительные различия в длине открытой рамки и аннотированного гена (рамка длиннее гена), вызванные тем, что открытая рамка начинается с нестандартного старт-кодона. Возможно,
алгоритм программы getorf старается найти самую длинную из возможных открытых рамок в данном месте генома. В этом случае в качестве старт-кодона использован кодон ATT. Голубым отмечен аннотированный
ген, красным - соответствующая ему открытая рамка.
Рисунок 8 (второй сверху). Еще один пример различия такого типа. Старт-кодон TTG.
Рисунок 9 (третий сверху). Старт-кодон CTG.
Рисунок 10 (самый нижний). Старт-кодон GTG. На рисунке 11 представлен участок генома с отмеченными рамкой и геном.
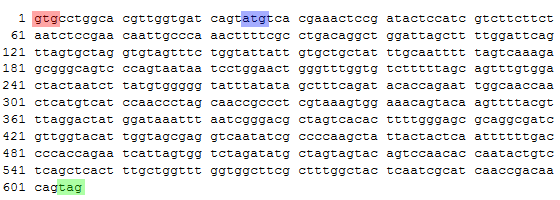
Рисунок 11. Участок последовательности, соответствующий найденной открытой рамке №13432. Красным отмечен старт-кодон этой рамки, GTG. Синим - обычный старт-кодон ATG, с которого начинается транкрипция гена
гипотетического белка (Nos7524_2187). Зеленым отмечен стоп-кодон, общий для гена и открытой рамки, - TAG.
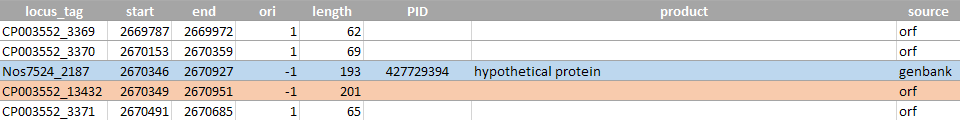
На рисунках 12-14 представлены пересекающиеся антипараллельные открытые рамки. Эти рамки расположены на прямой и на обратной цепи и перекрываются более чем на 150 п.о. Все примеры взяты из (примерно) первых 50000 п.о.
последовательности. Это позволяет предположить, что такое явление встречается в полном списке найденных открытых рамок достаточно часто.
Рисунок 12. Рамка №4 полностью находится внутри рамки №16769, перекрывание на 216 п.о. В окрестностях этих рамок нет аннотированных генов, сказать что-то конкретное сложно.
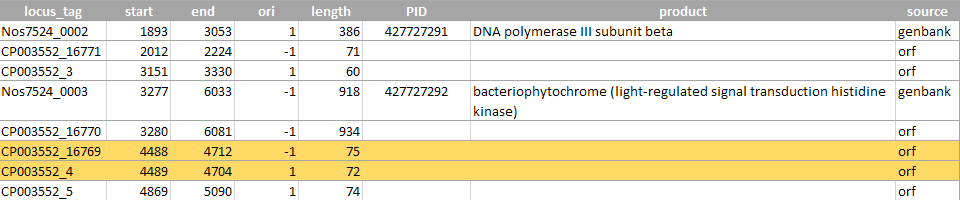
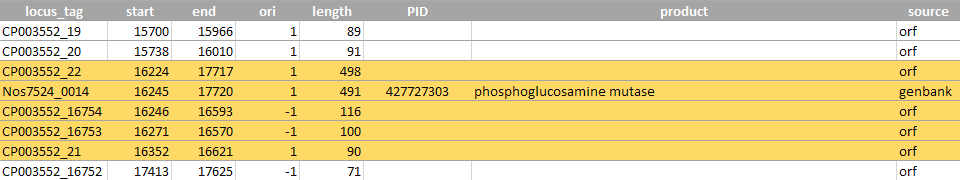
Рисунок 13. Рамки №16754 и №16753 полностью находятся внутри рамки №22. Также они перекрываются с рамкой №21 более чем на 150 п.о. В окрестностях этих рамок есть ген Nos7524_0014, которому соответствует
рамка №22. Возможно, транскрибируется именно эта рамка, так как она самая длинная, и это мешает использованию других открытых рамок.
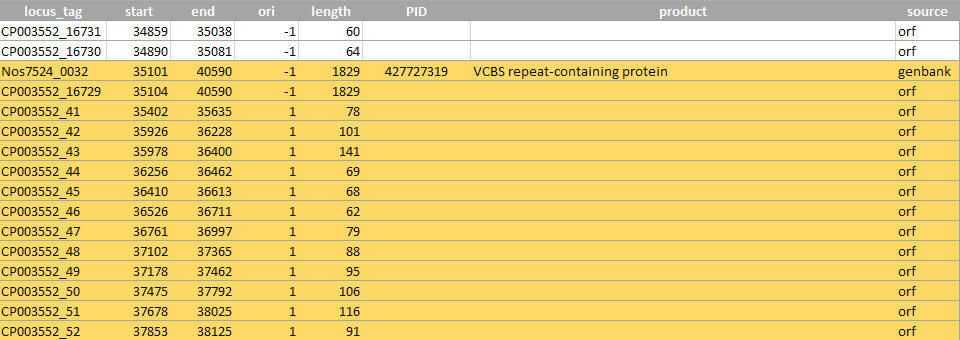
Рисунок 14. Рамки №41-57 (на рисунке представлены не все) полностью находятся внутри рамки №16729. Эта рамка очень длинная, при этом имеется аннотированный белок для этой рамки. Такой большой продукт,
скорее всего, предпочтительней множества маленьких цепей.
|