|
|
|
|
На данной странице выложены все задания блока 3. При нажатии на ссылку вы перемещаетесь в соответствующее место на странице.
Системы рестрикции-модификации в двух штаммах бактерии Citrobacter koseri
Для сравнения наборов систем рестрикции-модификации я использовала грамотрицательную бактерию Citrobacter koseri.
Геномы были взяты из базы данных NCBI (CP000822.1)
и из данных о секвенировании метагенома кишечника человека. Стоит отметить, что геном, лежащий в NCBI, был получен при секвенировании
образца, выделенного из мозга младенца, болеющего неонатальным менингитом.
Можно сделать следующие выводы:
Поиск последовательностей Шайна-Дальгарно в геноме бактерии Nostoc sp.
Последовательность Шайна-Дальгарно (SD) - участок связывания малой субъединицы прокариотической рибосомы с мРНК, обычно расположенный
за несколько нуклеотидов до начала трансляции. Считается, что у прокариот SD присутствует в начале большинства генов, и мутации в ее
консенсусной последовательности GGAGG или в комплементарном ей участке 16s рРНК приводят к подавлению
трансляции1.
Сначала с сервера NCBI я скачала сборку генома данной бактерии - кольцевую хромосому
CP003552.1. Затем были отобраны хорошие гены - достаточно длинные и с
адекватной аннотацией (с определенной точностью известен продукт белка). Так как геном Nostoc sp. большой (более 6Mb),
генов в нем также много - 5355, причем их длина варьирует от 90 до 30000 п.н. В результате была составлена выборка из 996 генов
(чтобы добиться нахождения достоверных мотивов). Для каждого гена с помощью скрипта был вырезан участок от -16 до -1 позиции до
начала кодирующей последовательности.
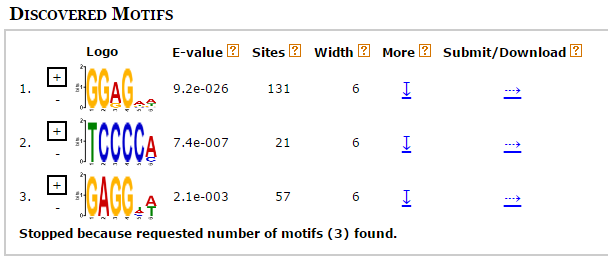
Рисунок 1. Три мотива, найденных программой MEME в промоторных областях генов бактерии Nostoc sp..
Как видно на рисунке 1, самый лучший мотив (с e-value 9.2e-026), значительно более достоверный, чем остальные, и является искомой
последовательностью SD.
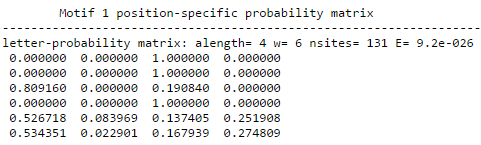
Рисунок 2. Позиционная весовая матрица (PWM) мотива Шайна-Дальгарно, построенная программой MEME. Рисунок 3. Лого найденной последовательности Шайна-Дальгарно.
С помощью программы FIMO я произвела поиск найденного мотива для всех остальных генов бактерии Nostoc sp. Для этого
были вырезаны участки от -26 до -1 позиции до начала кодирующей последовательности. Длина участков была увеличена, чтобы немного
снизить вероятность ошибок поиска для неправильно аннотированных генов.
Во-первых, были построены гистограммы распределения начала найденных SD от страта трансляции. Полученные изображения представлены на рисунках 4а-в. 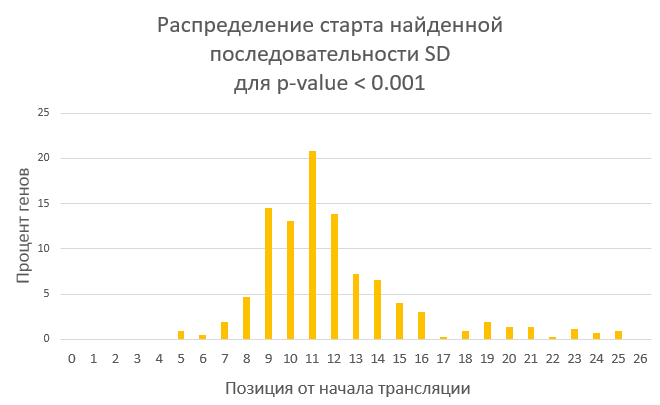
Рисунок 4а. Гистограмма распределения начала найденных SD от страта трансляции для порого p-value 0.001 (7,97% генов). Числа на оси X означают расстояние от начала SD до старта трансляции (по идее, их стоит рассматривать как отрицательные числа, так как SD расположена до начала кодирующей рамки). 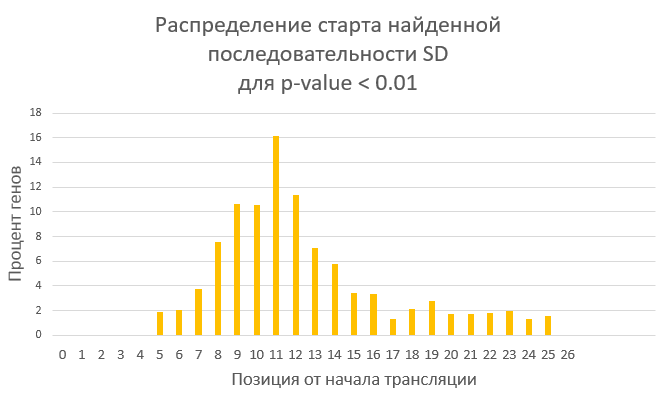
Рисунок 4б. Гистограмма распределения начала найденных SD от страта трансляции для порого p-value 0.01 (21,92% генов). 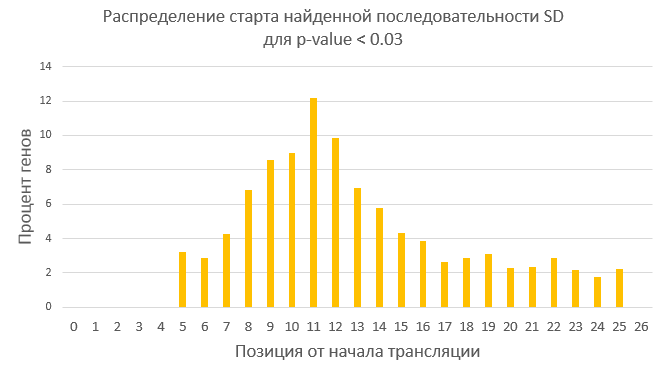
Рисунок 4в. Гистограмма распределения начала найденных SD от страта трансляции для порого p-value 0.03 (78,82% генов).
В основном старт SD приходится на -11 нуклеотид от начала трансляции (и конец, соответсвенно, на -7). Это в целом согласуется с
литературными данными, однако точно определить долю генов, содержащих SD, достаточно сложно. С одной стороны, полученные результаты
могут указывать и на высокую долю таких генов, и на относительно низкую, что может быть связано с нахождением случайных мотивов.
С другой стороны, литературные данные высказываются в пользу второго варианта (по крайней мере, генов, содержащих SD, должно
быть не больше половины).
Рисунок 5. Лого последовательности Шайна-Дальгарно, построенное по находкам этого мотива в геноме Nostoc sp. (для порога p-value 0.01). Изображение получено с помощью сайта LOGO.
Ссылки на использованную литературу:
Определение сайтов связывания транскрипционного фактора в участке хромосомы человекаДля начала полученные данные по ChIP-seq анализу были проверены с помощью программы FastQC. Результаты представлены по ссылке. График, отображающий качество нуклеотидов в чтениях, представлен на рисунке 6. Так как качество примерно одинаково по всей длине чтения и не сильно ухудшается к концу, а сами чтения достаточно короткие (36 нуклеотидов), обработка программой Trimmomatic не проводилась. 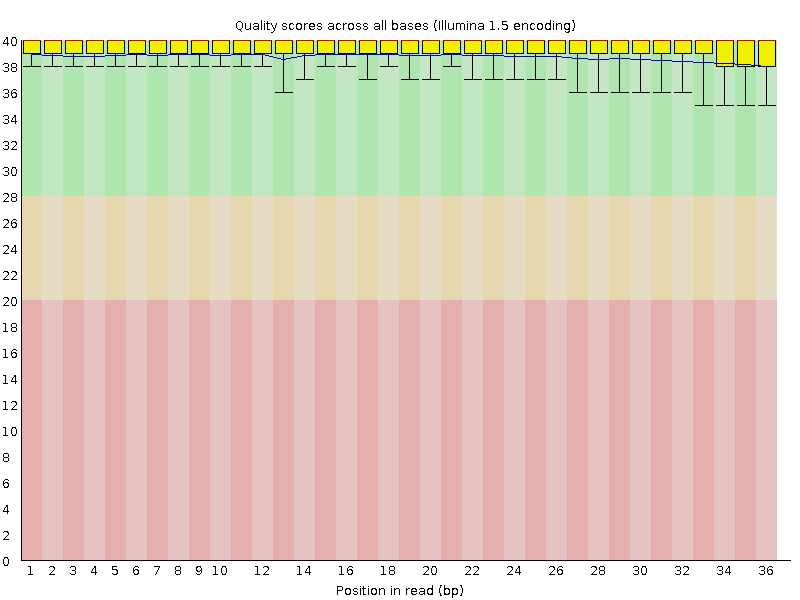
Рисунок 6. Распределение качества нуклеотида в риде по положению в риде. Иизображение получено с помощью анализа качества чтения программой FastQC.
Затем чтения были откартированы на геном человека hg19 (заранее проиндексированный) с помощью команды
bwa mem ../hg19/GRCh37.p13.genome.fa chipseq_chunk21.fastq > chipseq_chunk21.sam. Далее были использованы
следующие команды: samtools view -bSo chipseq_chunk21.bam chipseq_chunk21.sam (переводит выравнивание чтений
с референсным геномов в бинарный формат, с которым потом работают программы), samtools sort chipseq_chunk21.bam -T
chip_temp -o chipseq_chunk21.sorted.bam (сортирует выравнивание по координате начала чтения в референсе),
samtools index chipseq_chunk21.sorted.bam (индексирует отсортированный файл), samtools idxstats
chipseq_chunk21.sorted.bam > chipseq_chunk21.idxstats (записывает в файл
chipseq_chunk21.idxstats информацию о количестве чтений,
откартированных на каждый элемент генома) и samtools view -c chipseq_chunk21.sorted.bam (показывает,
сколько чтений в сумме было откартированно на все элементы генома).

Рисунок 7. Число чтений, откартированных на геном, по результатам работы описанных программ. 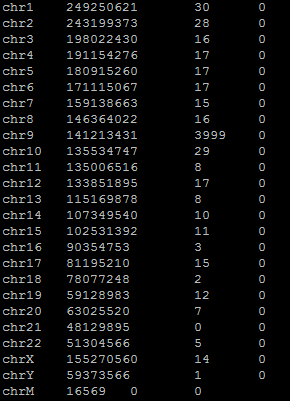
Рисунок 8. Распределение числа откартированных чтений по хромосомам. Больше всего чтений откартировано на 9 хромосому (3999 ридов, что составляет 93% всех ридов).
Далее я произвела поиск пиков с помощью программы MACS. Так как пиков было очень мало, использовалась команда
macs2 callpeak -t chipseq_chunkX.sorted.bam -n chipseq_chunk21 --nomodel. Были получены следующие файлы:
chipseq_chunk21_peaks.narrowPeak,
chipseq_chunk21_peaks.xls и
chipseq_chunk21_summits.bed.
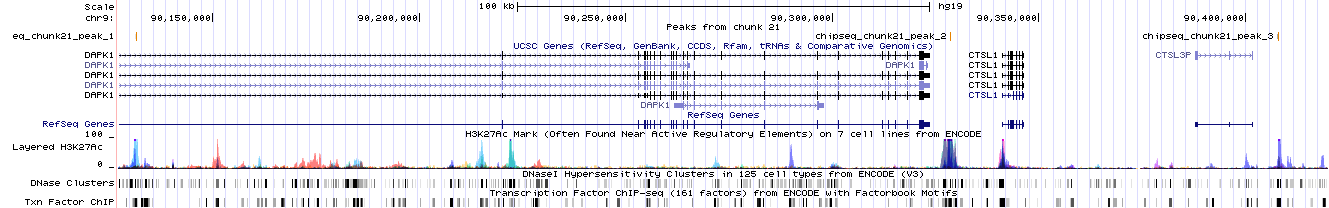
Рисунок 9. Расположение найденных пиков в геноме человека (сборка hg19). Представлен участок 9 хромосомы. Изображение получено с помощью геномного браузера UCSC. 
Рисунок 10. Увеличенный первый пик, занимающий позиции с 90,131,201 по 90,131,451 и имеющий длину 251 nt. 
Рисунок 11. Увеличенный второй пик, занимающий позиции с 90,328,381 по 90,328,707 и имеющий длину 327 nt. 
Рисунок 12. Увеличенный третий пик, занимающий позиции с 90,407,740 по 90,408,031 и имеющий длину 292 nt. Первый пик попадает внутрь гена DAPK1 (Death-associated protein kinase 1). Второй пик находится между генами DAPK1 и CTSL1 (Cathepsin L1). Третий пик расположен после гена CTSL3P (cathepsin L family member 3, pseudogene). Также для всех пиков я изучила их расположение относительно других известных транскрипционных факторов, данные для которых были получены в ходе других экспериментов и занесены в базу, которую можно визуализировать в геномном браузере. Результаты для 1, 2 и 3 пика представлены на риснуке 13. Можно заметить, что данный транскрипционный фактор колокализуется с другими факторами (также имеющими достаточно размытые сайты взаимодействия). К тому же, в области посадки этого фактора наблюдается изменение насыщенности метки H3K27ac. 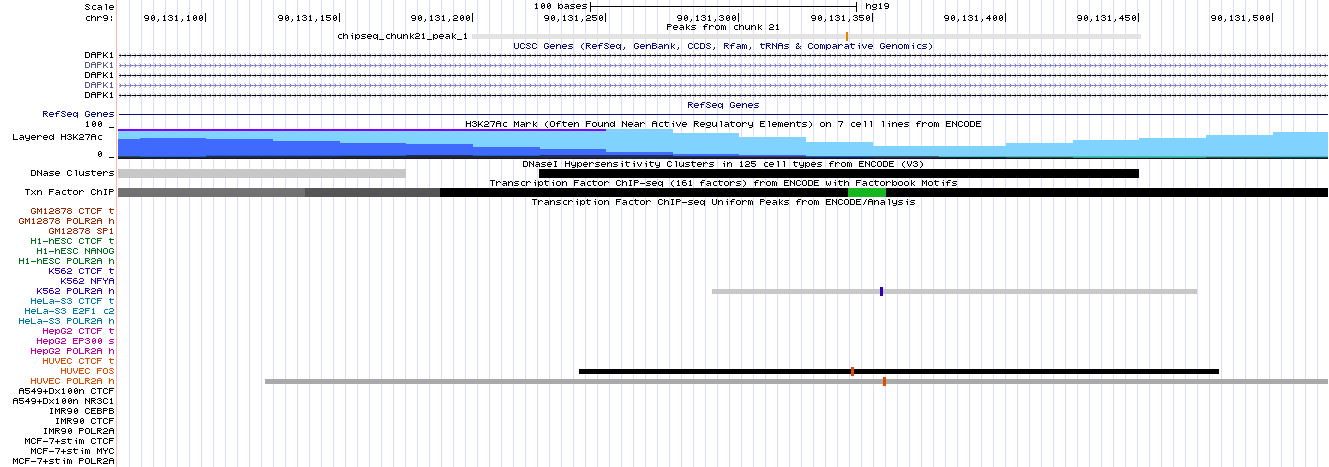
Рисунок 13а. Первый пик. Представлены данные по другим экспериментам для различных транскрипционных факторов. 
Рисунок 13б. Второй пик. 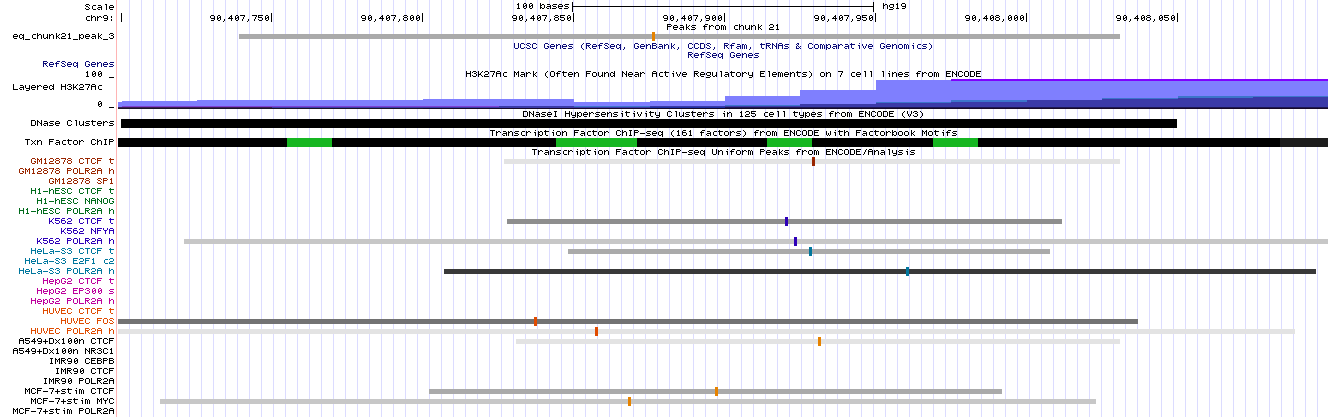
Рисунок 13в. Третий пик. Поиск сигналов TATA-бокс связывающего белка (TBP) в геноме человека
TBP является одним из ключевых ДНК-узнающих белков, необходимых для образования на промоторах генов комплекса TFIID и
инициации транскрипции с помощью Pol II. Тем не менее, лишь часть промоторов имеет сигнал TATA-box, связываемый TBP.
Консенсусная последовательность для связывания TBP - TATAWAAR.
На рисунке 14 представлен ген, перед которым нет сигнала TATA-box (все сигналы в окрестности промотора гена примерно одинаковы и могут быть рассмотрены как шум). Это ген ATP2B2, кодирующий АТФазу, транспортирующую Ca2+. Данный ген находится на 3 хромосоме (координаты 10,365,707 - 10,547,268, длина гена 181,562 п.о., он закодирован на (-)-цепи и содержит 23 экзона). На рисунке 14а показана вся промоторная область, на рисунке 14б - увеличенный участок, в котором должен был бы находиться сигнал TATA-box (с разрешением до отдельных нуклеотидов). 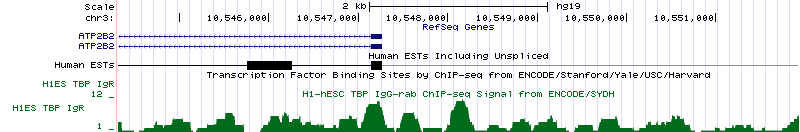
Рисунок 14а. Промоторная область гена ATP2B2. 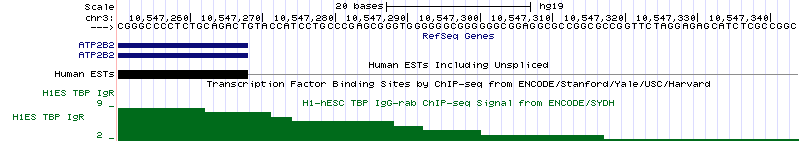
Рисунок 14б. Последовательность нуклеотидов в промоторной области гена ATP2B2. На рисунках 15а и 15б представлены аналогичные изображения промоторной области гена H2AFX, кодирующего один из вариантов второго гистона. Ген H2AFX расположен на 11 хромосоме (координаты 118,964,585 - 118,966,177, длина 1593 п.о., закодирован на (-)-цепи и содержит 1 экзон). 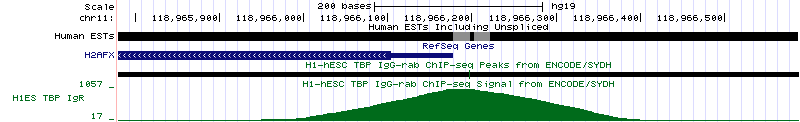
Рисунок 15а. Промоторная область гена H2AFX. 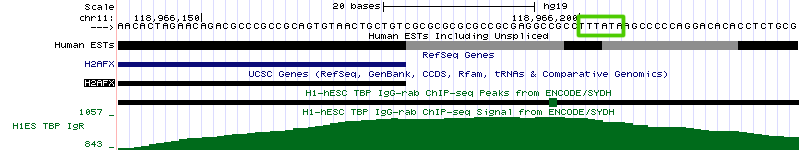
Рисунок 15б. Последовательность нуклеотидов в промоторной области гена H2AFX. Зеленой рамкой обведен сайт связывания TBP. Продукт гена DNMT1 участвует в метилировании цитозинов по 5 положению. Этот процесс необходим для регуляции уровней экспрессии генов, так как в промоторных областях многих генов находятся CpG-островки. Ген DNMT1 расположен на 19 хромосоме (координаты 10,244,022 - 10,305,755, длина 61,734 п.о., закодирован на (-)-цепи и содержит 40 экзонов). Его промоторная область представлена на рисунках 16а и 16б. 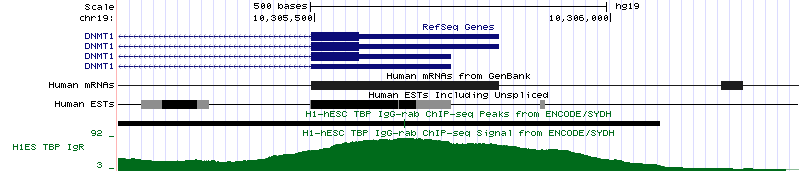
Рисунок 16а. Промоторная область гена DNMT1. 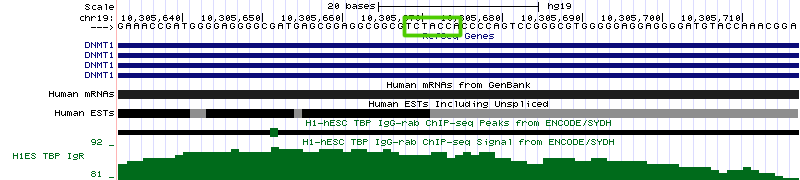
Рисунок 16б. Последовательность нуклеотидов в промоторной области гена DNMT1. Зеленой рамкой обведен сайт связывания TBP. Еще один ген, транскрипция которого зависит от TBP, - это CDC40, участвующий в регуляции прохождения клетки по клеточному циклу (рисунок 17). Расположен на 6 хромосоме (координаты 110,501,587 - 110,553,422, длина 51,836 п.о., закодирован на (+)-цепи и содержит 15 экзонов). 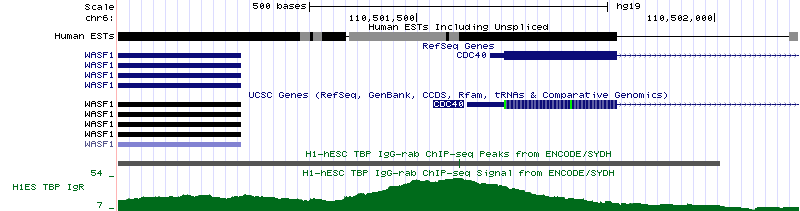
Рисунок 17а. Промоторная область гена CDC40. 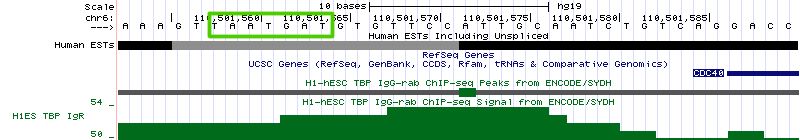
Рисунок 17б. Последовательность нуклеотидов в промоторной области гена CDC40. Зеленой рамкой обведен сайт связывания TBP. В двух из трех описанных случаев сигнал TATA-box расположен перед началом гена на расстоянии около 20 нуклеотидов. В третьем случае сайт связывания TBP расположен уже внутри самого гена. В целом, в геноме встречается расположение TATA-box и внутри, и перед геном, по крайней мере, по результатам данного анализа ChIP-seq. |
|
© Наталия Кашко, 2016 |