Совмещение структур
Первым заданием практикума было с помощью сервиса PDBeFold отобрать белки, являющиеся структурными гомологами выбранного нами белка(в мое случае - 4GUW). Это было сделано (выбраны структуры 3BWH, 4LGR, 1PUU и 3BJG), после чего c помощью этого же сервиса было проведено множественное выравнивание выбранных структур и 4GUW (результат работы PDBeFold - рис. 1.). На рис. 2. видно, что структуры совмещаются достаточно хорошо.
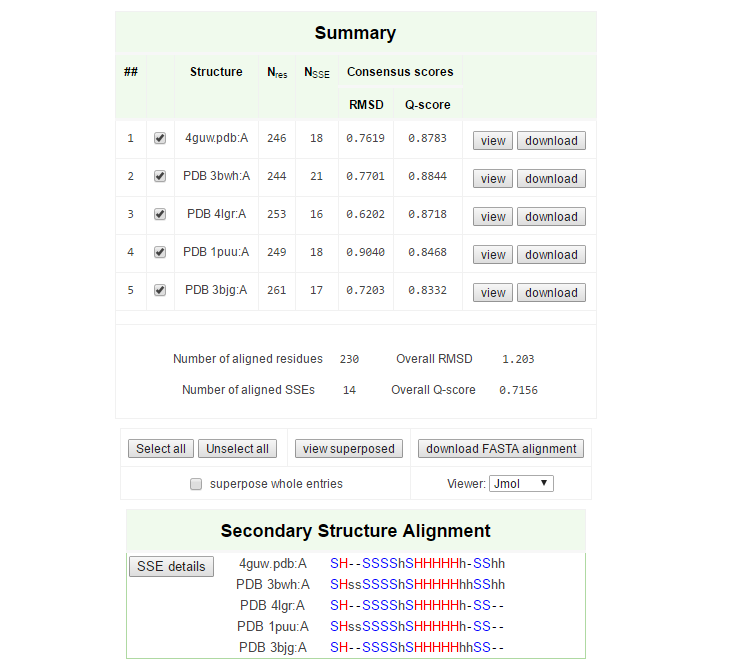
Рисунок 1. Результат работы множественного выравнивния на PDBeFold.
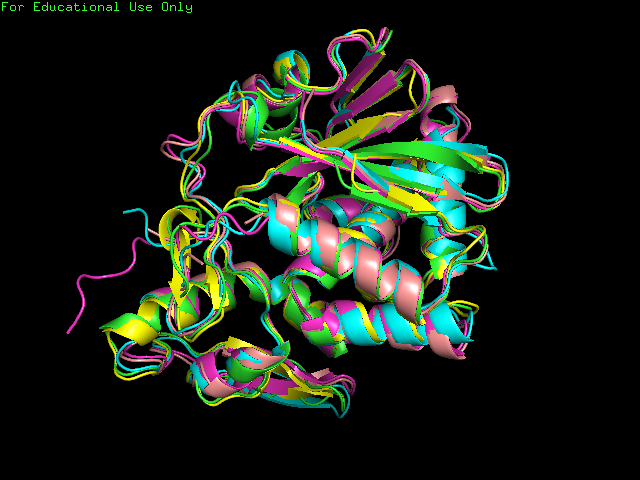
Рисунок 2. Выравнивание структур. Видно, что они совмещаются хорошо.
Из полученного выравнивания структур было получено выравнивание последовательностей. Кроме того, последовательности были выравнены с нуля с помощью сервиса T-Coffee. Было произведено сравнение полученных выравниваний и подробно разобраны три случая их несовпадения - таблица 1.. Файл сессии Pymol с выравнивание структур и выделенными разобранными несовпадениями можно скачать по этой ссылке. Файл проекта Jar с выравниваниями, полученными из структурного совмещения и с помощью сервиса T-Coffee можно скачать по этой ссылке. Файл с выраниванием, полученным на основе структурного совмещения можно скачать по этой ссылке. Файл с выраниванием, полученным с помощью T-Coffee можно скачать по этой ссылке.
Таблица 1. Разбор позиций, несовпадающих в выравнивании, полученном на основании структурного совмещения и в выравнивании, полученном с помощью T-Coffee.
| Выравненные аминокислоты (в структурном выравнивании) | Комментарий | Изображение, 4guw - розовый цвет, 3bwh - синий, 4lgr - оранжевый, 1puu - голубой, 3big - зеленый |
|---|---|---|
| 4guw:T28, 3bwh:K27, 4lgr:A34, 1puu:G31, 3big:G35 | В одной структуре (4lgr) на данном месте вообще нет остатка. Выравнивание же остальных остатков не позволяет с достаточно уверенностью утвержать, что они гомологичны. |  |
| 4guw:P86, 3bwh:V86, 4lgr:D94, 1puu:A92, 3big:D96 | Выравнивание отдельных частей, конечно, хорошо, но все равно не позволяет достоверно утверждать гомологичность остатков | 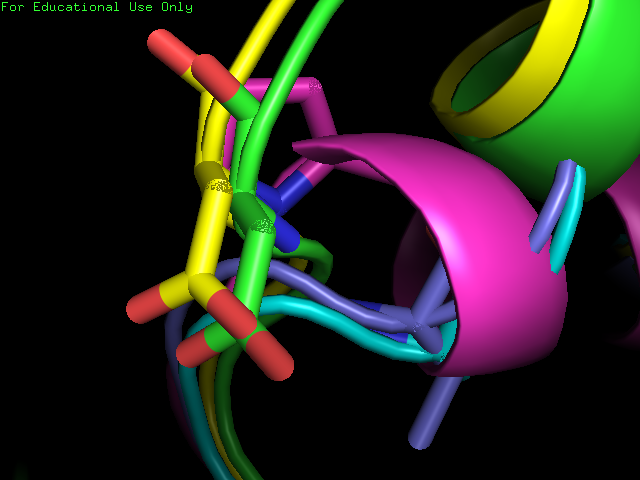 |
| 4guw:K102, 3bwh:Q100, 4lgr:R112, 1puu:R106, 3big:R110 | Можно уверенно сказать, что данные остатки гомологичны и в выравнивании должны быть в одной колонке. | 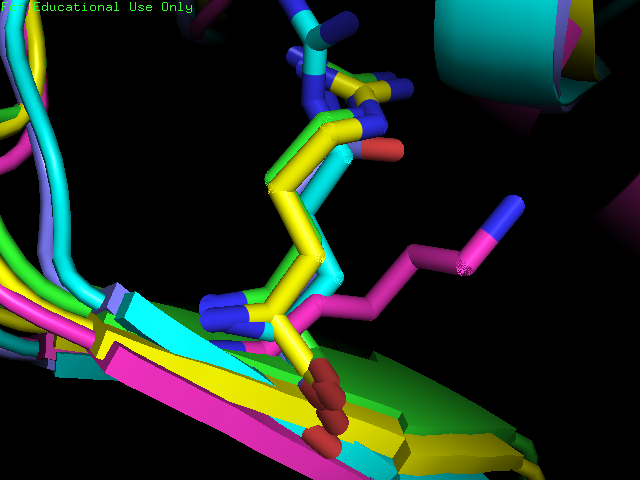 |
Следующим заданием было выбрать одну структуру константного домена T-клеточного рецептора из цепочки альфа и одну из - бета. Мною был взята структура 1QSE и из нее вырезаны альфа-домен из цепи D остатки 118-206, бета-домен - цепь E, остатки 119-246. Границы доменов были взяты из SCOP.
Далее с помощью сервиса SheeP были построены карты бета-дистов для цепочек алтфа и бета. Были выбраны соответствующие друг другу листы из цпочки бета и цепочки альфа ( рис.3 и рис.4). Далее были выбраны в этих листах консервативные цистеины (номера остатков 137 для альфа-цепи и 147 для бета, они оба образуют дисульфидную связь, информацию можно найти в PDB-файле).
Затем эти цистеины были выравнены, таким образом было получено выравнивание центрального тяжа. Итеративно за этим было построено выравнивание выбранных бета-листов. Была написана команда для PyMol, совмещающая структуры.
select good_align_alpha, resi 137-142+177-182+125-128+159-161 and alpha and name CA select good_align_beta, resi 145-150+191-196+128-131+173-175 and beta and name CA pair_fit good_align_alpha, good_align_betaЦеликом скрипт, строящий выраниваних этих структур можно скачать по это ссылке. Изображение хорошо выравненного участка приведено на рис.5 . Изображение, получаемое скриптом приведено на рис.6 .
Исходя из данного выравнивания можно сделать вывод и сходстве топологий двух данных структур, т.к все различающиеся участки находятся в одинаковых "ответвлениях" от тяжей. Стоит отметить, что программы поиска вторичных структур, находит в цепи бета моей структуры бета-лист, не аннотированный в PDB файле. Короткая проверка показала, что очень вероятно, что он действительно просто пропущен при аннотации. Потому для создания изображений использовался модифицированный файл с добавленной соответствующей аннотацией, его можно скачать по этой ссылке
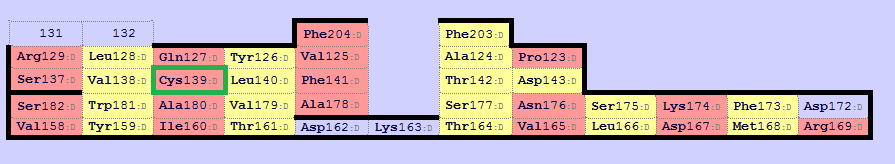
Рисунок 3. β-лист из цепи альфа. Консервативный цистеин выделен зеленым.
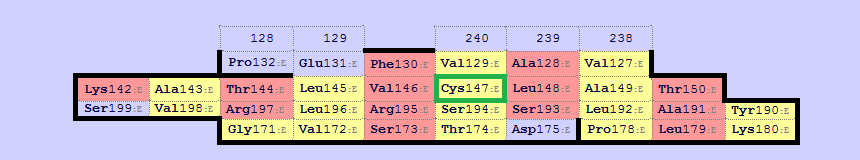
Рисунок 4. β-лист из цепи бета. Консервативный цистеин выделен зеленым.
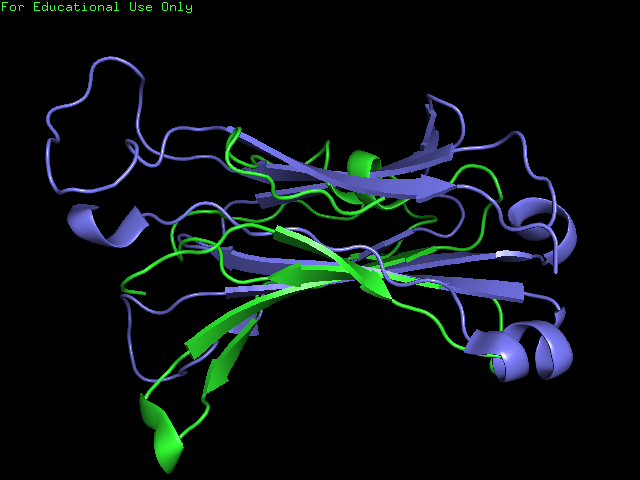
Рисунок 5. Изображение хорошо выравненных участков альфа и бета цепи
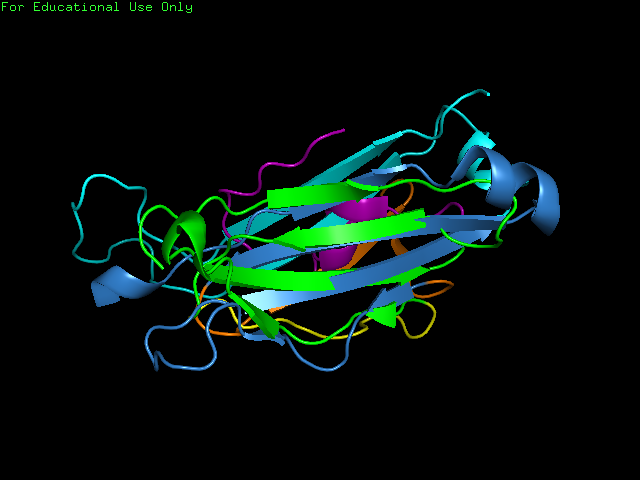
Рисунок 6. Изображение, получаемое скриптом.
Последним заданием данного практикума было нахождение пары белков, для которых гибкое структурное выравнивание включает существенно больше остатков, чем жесткое. Мною был взят пример из статьи, описывающей алгоритм FATCAT (Flexible structure alignment by chaining aligned fragment pairs allowing twists). Структура 1aj3 состоит из трех альфа-спиралей, а структура 2spcA - из трех. При этом отличие обуслено небольшой мутацией, которая переводит одну спирал в две, в остальном же структуры похожи. Но жесткое выравнивание (PDBeFold) этого не видит и в результате выравнивает куда меньше остатков ( рис. 7), FATCAT же выравнивает структуры целиком ( рис. 8).
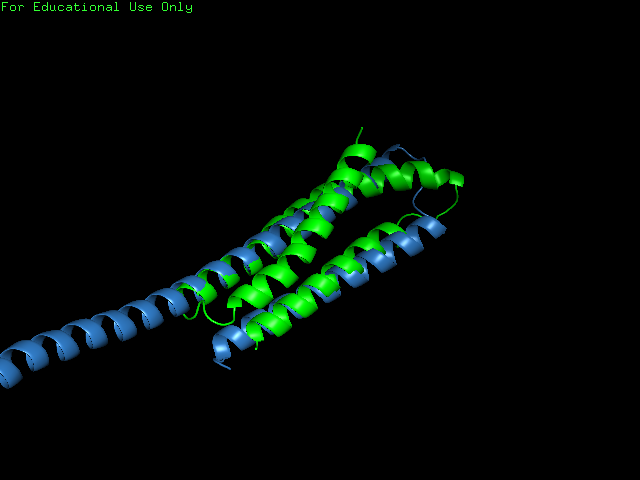
Рисунок 6. Жесткое выравнивание структур с помощью PDBeFold. 1aj3 (зеленый цвет) и
2spcA (синий цвет). Часть остатков не выравнена.
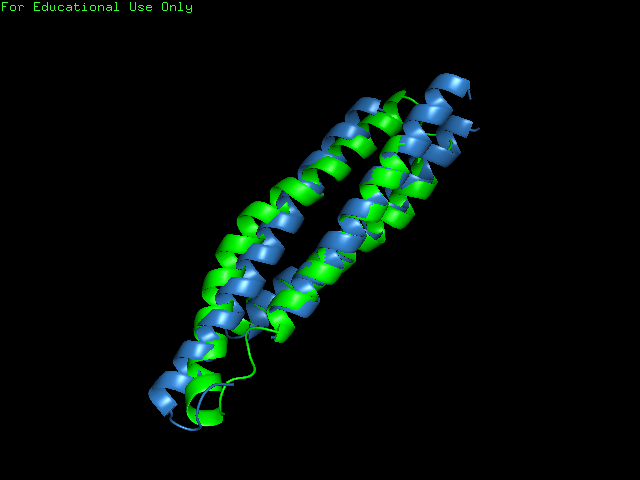
Рисунок 7. Гибкое выравнивание структур с помощью FATCAT. 1aj3 (зеленый цвет) и
2spcA (синий цвет). Структуры выравнены практически целиком.
