Ресеквенирование. Поиск полиморфизмов у человека
В данном задании требовалось найти и описать полиморфизмы у пациента. Задание выполнялось для чтений экзома пациента, которые нужно картировать на участок определенной хромосомы человека сборки генома hg19 (в данном случае использовалась хромосома 2).
Часть I: подготовка чтений
1. Анализ качества чтений.
Сначала было нужно проверить качество имеющихся чтений при помощи рограммы FastQC. Программа была запущена на kodomo командой fastqc chr2.fastq. Результатом работы программы является архив и файл в формате html, визуализирующий информцию о чтениях. На Рис. 1 представлен график качества чтений, сделанный программой FastQC до чистки. Синей линией на графике обозначено среднее качество чтений, центральными красными линиями обозначена медиана (значение качества, при котором половина чтений имеет качествоне выше указанного), желтыми прямоугольниками - интерквартальный размах (разница между верхней и нижней квартилями, диапазон значений качества, при котором качество 25% чтений на данной позиции выше нижней границы, а 75% - не выше верхней), черными "усами" обозначен размах мажду 10-й и 90-й процентилями (качество 10% чтений не выше нижней границы, 90% - не выше верхней). Поле графика разделено на 3 полосы зеленого, желтого и красного цветов, попадание в которые вышеперечисленных элементов графика позволяет судить о качестве чтений.
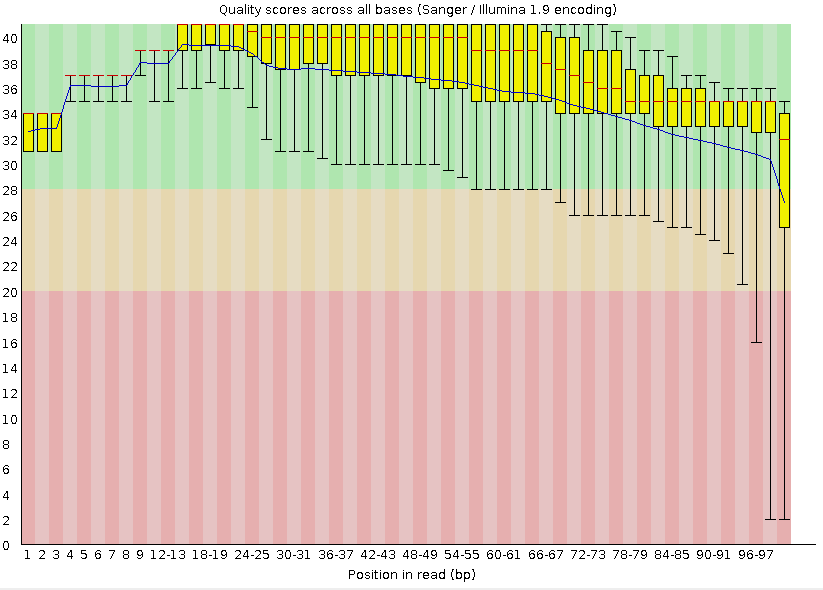
Рис. 1. "Per base sequence quality", график качества чтений, сделанный программой FastQC до чистки
2. Очистка чтений
Далее было необходимо очистить чтения припомощи программы Trimmomatic. Эта программа также была запущена с сервера kodomo командой java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr2.fastq outfile.fastq TRAILING:20 MINLEN:50. Первая опция отрезает с конца каждого чтения нуклеотиды с качеством ниже 20, вторая удаляет риды короче 50 нуклеотидов. До чистки было 10410 чтений, после - осталось 10191, было удалено 219 чтений.
Затем снова была запущена программа FastQC, чтобы узнать, какие чтения были удлены. Результатом снова стали архив и файл в формате html.
На Рис. 2 представлен график качества чтений, сделанный программой FastQC после очистки. Видно, что после очистки не стало
чтений, с "усами", заходящими в красную область поля графика, т.е. тех, у которых на концах были нуклеотиды плохого качества.
Теперь все позиции, кроме последней, расположены в зеленой области (последняя - в желтой), из чего можно заключить, что чистка
прошла успешно.
Еще один результат работы программы Trimmomatic состоял в удалении чтений с длиной меньше 50 нуклеотидов, теперь
длины чтений колеблются от 50 до 100 нуклеотидов (раньше: от 40 до 100).
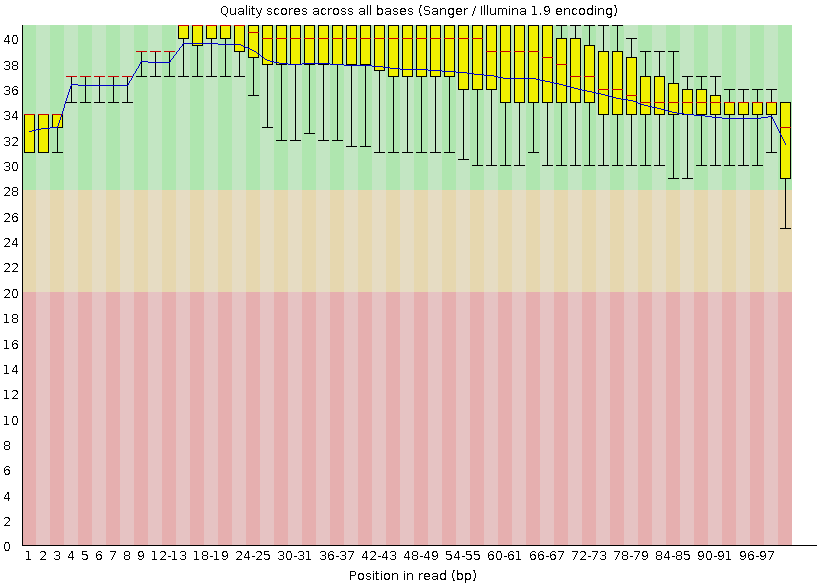
Рис. 2. "Per base sequence quality", график качества чтений, сделанный программой FastQC после чистки
Часть II: картирование чтений
3. Картирование чтений
На данном этапе надо было при помощи программы BWA откартировать очищенные
чтения. Для этого сперва была проиндексирована референсная последовательность хромосомы 2
(команда bwa index chr2.fasta).
После этого было построено выравнивание чтений и референсной последовательности в формате .sam
(команда bwa mem chr2.fasta outfile.fastq > align.sam).
4. Анализ выравнивания
Анализ был проведен при помощи пакета samtools. Для начала выравнивание чтений с референсной последовательностью было
переведено в формат .bam (команда samtools view -b -o align.bam align.sam).
Затем выравнивание в формате .bam было отсортировано по координате в референсе начала чтения
(команда samtools sort -T/tmp/align_sorted -o align_sorted.bam align.bam).
Далее полученный отсортированный файл был проиндексирован (команда samtools index align_sorted.bam).
После выполнения всех операций, описанных выше, была получена информация о количестве чтений, откартированных на геном
(команда samtools idxstats align_sorted.bam). Оказалось, что на геном откартировалось 10191 чтение - в точности
столько же, сколько остлось после очистки.
Часть III: анализ SNP
5. Поиск SNP и инделей
При выполнении данного блока заданий сперва был получен файл с полиморфизмами в формате .bcf
(команда samtools mpileup -uf chr2.fasta align_sorted.bam > snp.bcf).
Затем были определены отличия между референсом и чтениями (команда bcftools call -cv snp.bcf > snp.vcf).
Всего было найдено 80 полиморфизмов, из них 9 инделей и 71 замена. Три из них были выбраны и описаны в Таблице 1.
| Таблица 1. Описание трех полиморфизмов | |||||
|---|---|---|---|---|---|
| Координата | Тип полиморфизма | Было в референсе | Найдено в чтениях | Глубина покрытия данного места | Качество чтений в данном месте |
| 55516323 | Замена | T | G | 112 | 225.009 |
| 238428823 | Замена | А | Т | 2 | 40.7649 |
| 238453824 | Индель | TGGAGG | TGG | 2 | 15.7046 |
У первого из описанных полиморфизмов хорошие покрытие и качество, у других двух - плохие.
6. Аннотация SNP
Здесь требовалось проаннотировать только полученные snp при помощи программы annovar по БД refgene, dbsnp, 1000genomes,
GWAS, Clinvar.
Программа работает с файлами в формате avinput,которые можно получить из файла .vcf с помощью скрипта convert2annovar.pl
(команда perl convert2annovar.pl -format vcf4 /nfs/srv/databases/ngs/akmarina/snp.vcf >
/nfs/srv/databases/ngs/akmarina/snp.avinput).
Чтобы узнать, какие из из найденных замен имеют rs (идентификатор в базе данных однонуклеотидных полиморфизмов; уже изучены
или известны), была выполнена команда perl annotate_variation.pl -filter -out /nfs/srv/databases/ngs/akmarina/rs
-build hg19 -dbtype snp138 /nfs/srv/databases/ngs/akmarina/snp.avinput humandb/.
Далее были проведены аннотирования по БД, указанным выше. Команды:
dbnsp: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype snp138 -buildver hg19 snp.avinput -outfile dbs /nfs/srv/databases/annovar/humandb/ 1000genomes: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter snp.avinput -buildver hg19 /nfs/srv/databases/annovar/humandb/ -dbtype 1000g2014oct_all -out 1000g GWAS: perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno snp.avinput -build hg19 -dbtype gwasCatalog /nfs/srv/databases/annovar/humandb/ -out gwas Clinvar: perl /nfs/srv/databases/annovar/annotate_variation.pl snp.avinput /nfs/srv/databases/annovar/humandb/ -filter -dbtype clinvar_20150629 -buildver hg19 -out clinvar Refgene: perl /nfs/srv/databases/annovar/annotate_variation.pl -out refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/
Из всех SNP 70 имеют rs, 10 не имеют. Наименьшая частота SNP = 0.0227636, наибольшая частота = 0.994209. (ссылка).
В клинической аннотации SNP представлено 4 замены. Первая влияет на рост, вторая и третья вызывают болезнь Крона, а четвертая - рак простаты. (ссылка).
В БД RefSeq SNP подразделяется на exonic (variant overlaps a coding), splicing (variant is within 2-bp of a splicing junction), ncRNA (variant overlaps a transcript without coding annotation in the gene definition), UTR5 (variant overlaps a 5' untranslated region), UTR3 (variant overlaps a 3' untranslated region), intronic (variant overlaps n intron), upstream (variant overlaps 1-kb region upstream of transcription start site), downstream (variant overlaps 1-kb region downstream of transcription start site), intergenic (variant is in integric region).
БД RefSeq выделила 35 het замен и 43 hom. Из них в UTR3 попало 7 замен, в intronic - 62, в UTR5 - 2, в exonic - 7 и в ncRNA - одна замена. Замены затронули гены CCDC88A (UTR3, intronic SNP), ATG16L1 (UTR5, intronic, UTR3, exonic SNP) и MLPH (intronic, exonic, ncRNA SNP).