Построение дерева по нуклеотидным последовательностям
В задании требовалось построить филогенетическое дерево выбранных бактерий, используя последовательности РНК малой субъединицы рибосомы (16S rRNA). Искомые последовательности были добыты из *.frn файлов, находящихся в базе полных геномов NCBI. Сводная информация о выбранных геномах представлена в Таблице 1.
| Таблица 1. Отобранные представители бактерий | ||
|---|---|---|
| Название | Мнемоника | Штамм |
| Bacillus anthracis | BACAN | A0248 |
| Clostridium tetani | CLOTE | 12124569 |
| Enterococcus faecalis | ENTFA | 62 |
| Finegoldia magna | FINM2 | ATCC 29328 |
| Geobacillus kaustophilus | GEOKA | HTA426 |
| Lactobacillus acidophilus | LACAC | La 14 |
| Staphylococcus aureus | STAAR | JH9 |
| Streptococcus pneumoniae serotype 4 | STRPN | AP200 |
В каждом из файлов с расширением .frn было несколько последовательностей 16s-рРНК (копии гена). Выбранные последовательности были собраны в единый
файл в fasta-формате (файл можно скачать по ссылке). Названием каждой последовательности является мнемоника соответствующей
бактерии.
Выравнивание было построено при помощи сервера MUSCLE.
Файл с выровненными последовательностями доступен по ссылке.
Файл с выравниванием был импортирован в программу MEGA методом "Analyze", после чего по нему было реконструировано дерево методом Maximum Likelihood. Результат работы программы представлен на Рис. 1.

Рис 1. Изображение дерева, реконструированного программой MEGA методом Maximum Likelihood.
Реконструированное дерево отличается от правильного. Из всех нетривиальных ветвей только ветви {CLOTE, FINM2}, {STRPN, ENTFA} и {(STRPN, ENTFA), LACAC} были реконструированы правильно. Соответственно, поскольку всего у данного дерева 5 нетривиальных ветвей, то 2 ветви были реконструированы неверно.
{kind=link}
Построение и анализ дерева, содержащего паралоги
В этом задании надо было найти в своих бактериях достоверные гомологи белка CLPX_BACSU.
были использованы файлы, лежащие в директории P:\y15\term4\Proteomes (они содержат скачанные из Uniprot полные протеомы выбранных бактерий).
Командой cat file1 >> file2 файлы были соединены вместе в один файл. Затем был проведен поиск гомологов программой blastp с входной
последовательностью данного белка по базе данных - полученному файлу с протеомами. При поиске был взят порог E-value 0,001. При поиске были использованы
следующие команды:
- makeblastdb -in bacs4db.fasta -dbtype prot -out db.fasta
- blastp -query CLPX_BACSU.fasta -evalue 0.001 -db db.fasta -outfmt 6 -out blastres.fasta
В выдаче blastp всего 45 находок, из них 6 принадлежат белку CLPX различных бактерий. В выдаче также 10 находок - по 2 находки, принадлежащие белку HSLU, на каждую из бактерий BACAN, ENTFA, GEOKA, LACAC, STAAR. Остальные находки представлены в единственном экземпляре.
Для дальнейшей работы нужно было из всех последовательностей в файле с протеомами выбрать только нужные 45. Для этого был написан скрипт, отбирающий последовательности по их ID в файле с находками blast'a и записывающий их в новый файл. Полученный файл был импортирован в программу MEGA, затем его последовательности были выровнены при помощи Muscle. По выравниванию методом Maximum Likelihood было построено филогенетическое дерево (Рис. 2).
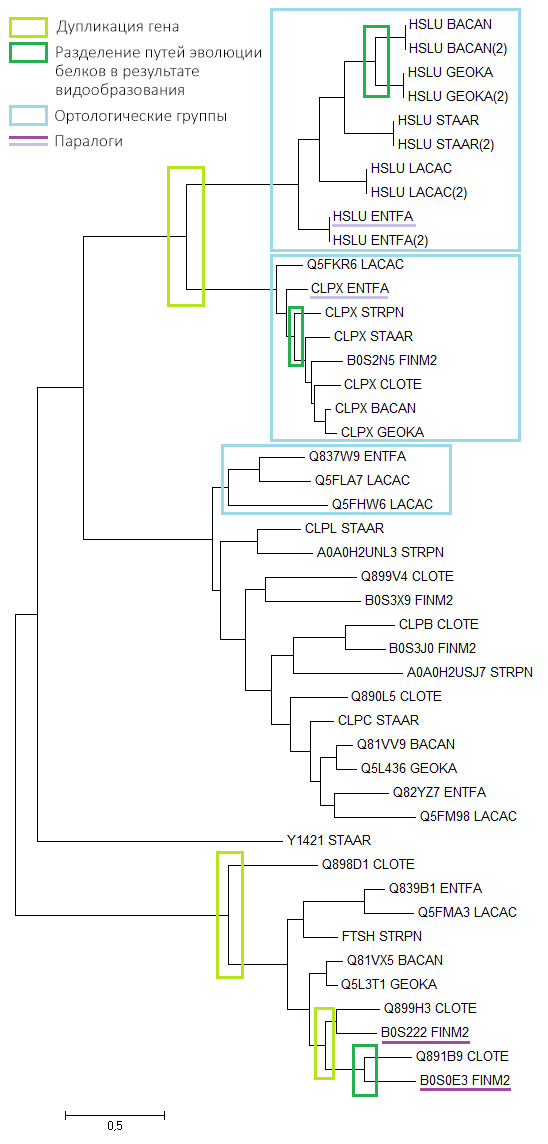
Рис 2. Филогенетическое дерево последовательностей гомологов белка CLPX_BACSU, реконструированное методом Maximum Likelihood в программе MEGA.
На полученном дереве были выделены следующие эволюционные события (при предположении, что дерево было реконструировано верно):
- Дупликация гена: два разных белка выполняют разные функции, но присутствуют у всех/почти всех видов. Например, белки CLPX и HSLU представляют собой разные субъединицы АТФ-зависимой протеазы и оба встречаются почти у всех видов.
- Разделение путей эволюции белков в результате видообразования: такое разделение можно считать выделением групп ортологов.
Также на дереве были указаны:
- Ортологические группы: два гомологичных белка будем называть ортологами, если они: а) из разных организмов; б) разделение их общего предка на линии, ведущие к ним, произошло в результате видообразования. Рамками выделены группы, внутри которых белки попарно являются ортологами.
- Паралоги: два гомологичных белка из одного организма.