Домены и профили
Выбор домена и архитектуры
В Pfam я выбрал DNA ligase C-terminal домен (AC PF17879, 1 белок в seed, 161 - в full, длина профиля HMM 109 аминокислота) и двухдоменную архитектуру
DNLI_BPT7(AC P00969) с DNA_ligase_A_M(AC PF01068). Порядок доменов DNA_ligase_A_M - DNA_ligase_C-terminal, белков с данной архитектурой 77.
Скачал последовательности всех белков с С-концевой ДНК лигазой в fasta-файл.
С помощью скрипта перенес оттуда AC в файл.
Список белков, содержащих двухдоменную структуру с DNA ligase C-terminal получил в разделе Architectures.
Выравнивание белков с двухдоменной архитектурой.
С помощью скрипта получил fasta-файл с последовательностями белков, содержащих выбранную архитектуру. В JalView алгоритмом muscle with defaults получил следующее выравнивание. Проверил координаты доменов в белках на соответствующих страницах выравниваний, удалил небольшой кусок вначале выравнивания. Далее удалил часть последовательностей, создающих большие гэпы в остальных, а так же убрал высокосходные последовательности на 93% уровне, так чтобы осталось чуть больше половины белков(39/77). Новое выравниваниеHMM профиль.
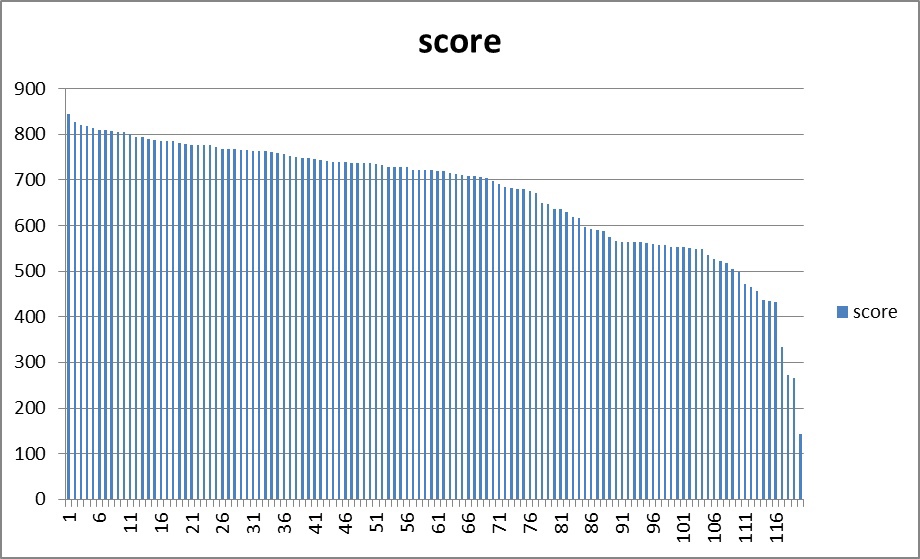
При помощи команд(см. ниже) я построил HMM-профиль данной архитектуры длины 385 аминокислот, откалибровал его и произвел поиск данной архитектуры по файлу со всеми белками семейства.hmm2build profile alnrev.fasta hmm2calibrate profile hmmsearch --cpu=1 profile full.fasta > hmm.txt #click hereВо всех последовательностях с низкими значениями e-value была найдена архтектура. С помощью средств Microsoft Excel 2010 и файлов, полученных ранее (включая табулированную часть файла находок и "чистые" АС белков с архитектурой), составлена электронная таблица, содержащая информацию о последовательностях, в которых проводился поиск домена(AC, найдена ли архитектура, участвовала ли в потроении профиля, был ли найден hmm2search, score, E-value, длина). Excel-full, txt-main. При подготовке таблицы выяснилось что в full.fasta некоторые последовательности повторяются. Дубликаты появились только в находках и в списке всех АС генов с лигазным доменом, я их удалил. Таким образом, уникальных АС 120(и последовательностей тоже), а не 161, как указано в full в Pfam. Чтобы получить порог, дающий наименьшее значение параметра F1, и построить ROC кривую, был использован ещё один скрипт, любезно предоставленный Владиславом Мурзиным. Значение порогового веса с наибольшим значением F1 составило 434,8. Более менее линейная форма графика объясняется плавным изменением веса.
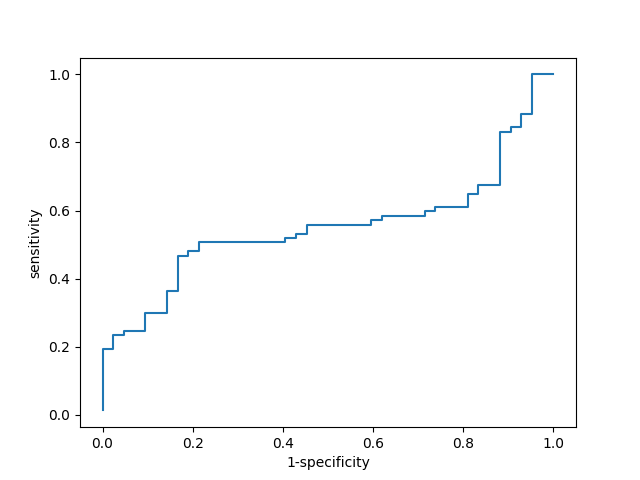
Рис. 1. ROC-кривая.