| Учебный сайт Саши Погорельской |

|
|
||||||
| Главная | Семестры | Скрипты | Обо мне | Ссылки | ||
|
|
Построение парного выравнивания Для выполнения практикума используется множественное выравнивание и PDB-файл с совмещением 2 структур. Это структуры двух последних белков, при удалении других белков и пустых колонок получается файл с парным выравниванием. Графическая интерпретация исходного множественного и парного выравниваний сохранена в проекте Jalview.
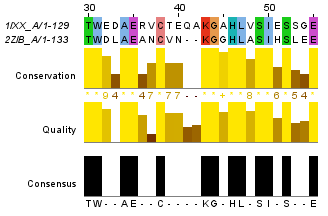
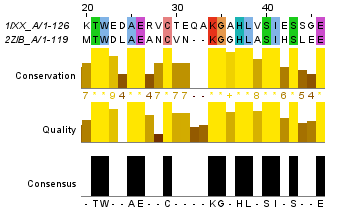
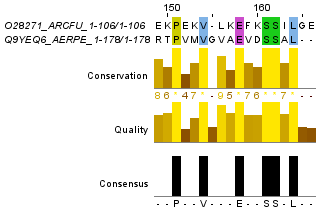
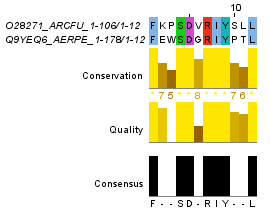
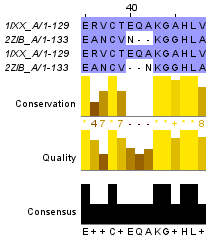
Для построения глобального выравнивания, часть которого изображена на рисунке 1, я использовала программу needle из пакета EMBOSS. Исходными файлами для работы этой программы являются 2 файла в формате fasta с аминокислотными последовательностями. По умолчанию выходной файл выглядит так. Для сохранения файла в формате fasta используется опция -aform fasta (такой выходной файл). Аналогично можно сделать локальное выравнивание, с помощью программы water. Здесь можно скачать файл с выравниваем по умолчанию, а здесь - в формате fasta. На рисунках 1 и 2 приведены одинаковые фрагменты последовательностей, различаются только номера остатков. Есть скачать проект с этими двумя выравниваниями, то можно легко проверить полное совпадение выравниваний. Так как локальное выравнивание не обязано распространяться на всю последовательность, концевые фрагменты отрезаны. Теперь проведем аналгоичные действия с заведомо негомологичными последовательностями. Для глобального выравнивания: файл с описанием выравнивания, в формате fasta. Те же файлы для локального выравнивания: текстовый и fasta. Частично эти выравнивания изображены на рисунках 3 и 4. Глобальное выравнивание содержит очень много гепов, фрагменты сходства короткие. Характеристики всех этих выравниваний получены с помощью функции infoalign (EMBOSS). Наиболее значимые сведения представлены в таблице 1. В колонке SeqLen - длина последовательности, AlignLen - длина выравнивания без учета гепов на конце. Количество открытий гепов указано в колонке Gaps, а их общая длина - GapLen. В колонке Ident таблицы 1 для первой последовательности указывается количество всех аминокислотных остатков, а для второй - количество полностью совпадающих. Similar - сходные остатки. Для гомологичных последовательностей лучшим локальным выравниванием является глобальное (без гепов на конце), для заведомо негомологичных - лишь небольшой участок длиной в 12 остатков. Таблица 1. Сведения о полученных выравниваниях
Полностью результаты можно посмотреть, если скачать исходный проект. Для проверки правильности выравнивания (то есть вероятности гомологии) можно использовать также пространственную структуру белков. Гомологичные белки обладают сходными функциями и, значит, сходным строением. С помощью программы SupCheck можно получить скрипт, который последовательно выделяет колонки выравнивания при запуске в RasMol. Для выравниваний, полученных изначально и с помощью needle, я проанализировала соответствие пространственных структур. В проекте Jalview в строках Correct alignment совмещенные остатки отмечены буквой "S". Колонки, которые я считаю выравненными достаточно достоверно, отмечены знаком "+". Рассмотрим возникшие ошибки. Ошибки первого рода: аминокислотные остатки отмечены "+", но не совмещены. Ошибки второго рода: остатки совпадают в структуре, но в разных колонках или не отмечены "+". Для исходного выравнивания найдено 16 ошибок первого рода и 26 ошибок второго рода. Для глобального выравнивания (needle) 18 - первого рода и 29 - второго. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| © Pogorelskaya Sasha | Last modification date: 19.02.15 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||